Lama Alzoebie, Dong Li, Xiang Wang, David R Weber, Michael A Levine
{"title":"Unusual <i>PHEX</i> variants implicate uncommon genetic mechanisms for X-linked hypophosphatemic rickets.","authors":"Lama Alzoebie, Dong Li, Xiang Wang, David R Weber, Michael A Levine","doi":"10.1093/jbmrpl/ziae152","DOIUrl":null,"url":null,"abstract":"<p><p>X-linked hypophosphatemic rickets (XLH), the most common form of hereditary rickets, is characterized by renal phosphate wasting and abnormal vitamin D metabolism due to elevated circulating levels of the phosphatonin fibroblast growth factor 23 (FGF23). Dominant inactivating variants of the phosphate regulating endopeptidase homolog, X-linked (<i>PHEX</i>), gene are present in patients with XLH, and more than half of affected patients carry de novo variants. We report on 3 families in whom affected members had highly unusual <i>PHEX</i> pathogenic variants. In 1 family we identified a previously described deep intronic <i>PHEX</i> variant (c.1768 + 173A>G) in the proband and her affected son. This variant is also near a previously reported PHEX variant (c.1768 + 177_1768 + 180dupGTAA) and is predicted to affect splicing by SpliceAI (delta score: 0.95) through creation of a new donor splice site. In a second proband we identified 2 pathogenic de novo and novel <i>PHEX</i> variants, c.2083delT (p.Ser695Profs*45) and c.2085delC (p.Tyr696Thrfs*44), that were present on different alleles, consistent with mosaicism for 3 <i>PHEX</i> alleles. The third proband also carried 2 <i>PHEX</i> variants (c.755 T>C [p.Phe252Ser] and c.759G>A [p.Met253Ile]), but in this case both variants were present on the same <i>PHEX</i> allele. These studies expand the molecular catalog of pathogenic <i>PHEX</i> variants in XLH and emphasize the importance of deep intronic sequencing and comprehensive family studies. Conventional approaches to genetic diagnosis may not be adequate to identify or characterize the disease-causing variants in the <i>PHEX</i> gene in some patients with likely XLH.</p>","PeriodicalId":14611,"journal":{"name":"JBMR Plus","volume":"9 1","pages":"ziae152"},"PeriodicalIF":2.4000,"publicationDate":"2024-11-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11646311/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"JBMR Plus","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1093/jbmrpl/ziae152","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2025/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"ENDOCRINOLOGY & METABOLISM","Score":null,"Total":0}
引用次数: 0
Abstract
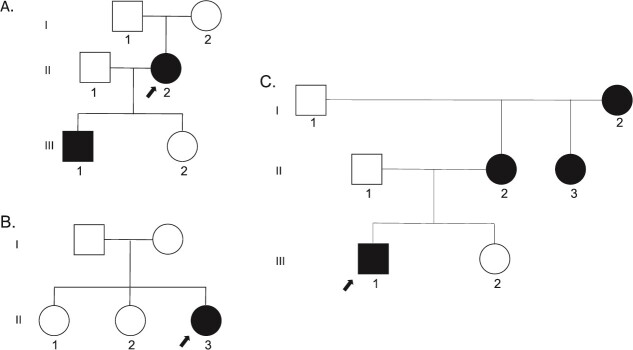
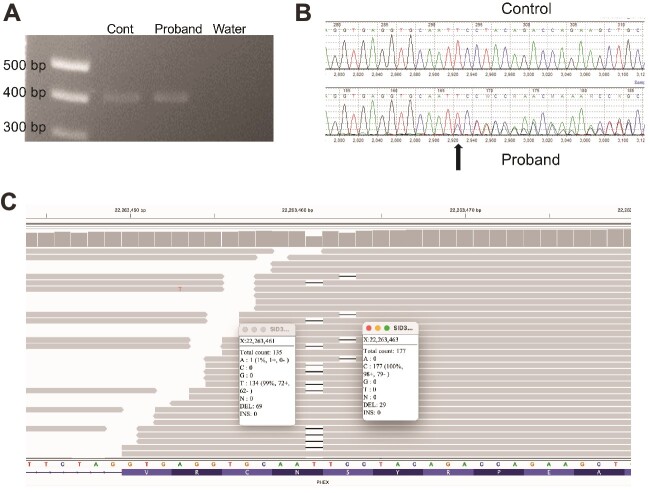
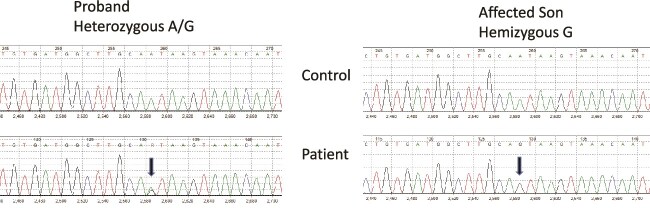
X-linked hypophosphatemic rickets (XLH), the most common form of hereditary rickets, is characterized by renal phosphate wasting and abnormal vitamin D metabolism due to elevated circulating levels of the phosphatonin fibroblast growth factor 23 (FGF23). Dominant inactivating variants of the phosphate regulating endopeptidase homolog, X-linked (PHEX), gene are present in patients with XLH, and more than half of affected patients carry de novo variants. We report on 3 families in whom affected members had highly unusual PHEX pathogenic variants. In 1 family we identified a previously described deep intronic PHEX variant (c.1768 + 173A>G) in the proband and her affected son. This variant is also near a previously reported PHEX variant (c.1768 + 177_1768 + 180dupGTAA) and is predicted to affect splicing by SpliceAI (delta score: 0.95) through creation of a new donor splice site. In a second proband we identified 2 pathogenic de novo and novel PHEX variants, c.2083delT (p.Ser695Profs*45) and c.2085delC (p.Tyr696Thrfs*44), that were present on different alleles, consistent with mosaicism for 3 PHEX alleles. The third proband also carried 2 PHEX variants (c.755 T>C [p.Phe252Ser] and c.759G>A [p.Met253Ile]), but in this case both variants were present on the same PHEX allele. These studies expand the molecular catalog of pathogenic PHEX variants in XLH and emphasize the importance of deep intronic sequencing and comprehensive family studies. Conventional approaches to genetic diagnosis may not be adequate to identify or characterize the disease-causing variants in the PHEX gene in some patients with likely XLH.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


