The Influence of the Solvation on the Bonding of Molecular Complexes of Diatomic Halogens With Nitrogen-Containing Donors and Their Stability With Respect to the Heterolytic Halogen-Halogen Bond Splitting
Anna V. Pomogaeva, Anna S. Lisovenko, Alexey Y. Timoshkin
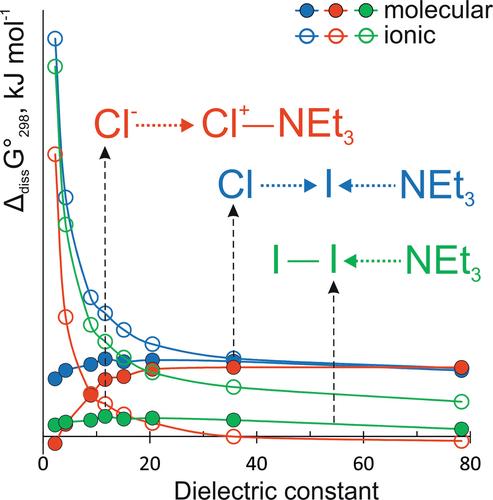
{"title":"The Influence of the Solvation on the Bonding of Molecular Complexes of Diatomic Halogens With Nitrogen-Containing Donors and Their Stability With Respect to the Heterolytic Halogen-Halogen Bond Splitting","authors":"Anna V. Pomogaeva, Anna S. Lisovenko, Alexey Y. Timoshkin","doi":"10.1002/jcc.27549","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>In the framework of SMD approach a systematic computational study of structural, electronic and thermodynamic properties of molecular complexes of Cl<sub>2</sub>, ICl and I<sub>2</sub> with series of N-containing Lewis bases in solvents of different polarity was carried out. Results indicate that molecular complexes of Cl<sub>2</sub> with strong and medium-strong LB undergo spontaneous ionization in the acetonitrile solution. The increase of the solvent polarity can change the nature of interaction in X'X<span></span>LB systems from molecular X'X ← LB donor-acceptor complexes to 3-center 4-electron bound X'<span></span>→X<sup>+</sup> ← LB in solvents of medium polarity and to the contact ion pairs X'<span></span>→[X<span></span>LB]<sup>+</sup> in polar solvents. Thus, the controlled generation of cationic [LB∙X]<sup>+</sup> species is possible by varying the nature of LB, varying the nature of the solvent, and varying the nature of the halogen X. Molecular Cl<sub>2</sub> has the greatest tendency to form ionic species in polar solvents. Spontaneous ionization of molecular nσ complexes of chlorine with strong LB in medium-polar solvents (starting from OEt<sub>2</sub>, <i>ε</i> = 4.24) should not be neglected and single point solvation energy computations on gas phase optimized geometries are not reliable for such systems.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-12-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27549","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
In the framework of SMD approach a systematic computational study of structural, electronic and thermodynamic properties of molecular complexes of Cl2, ICl and I2 with series of N-containing Lewis bases in solvents of different polarity was carried out. Results indicate that molecular complexes of Cl2 with strong and medium-strong LB undergo spontaneous ionization in the acetonitrile solution. The increase of the solvent polarity can change the nature of interaction in X'XLB systems from molecular X'X ← LB donor-acceptor complexes to 3-center 4-electron bound X'→X+ ← LB in solvents of medium polarity and to the contact ion pairs X'→[XLB]+ in polar solvents. Thus, the controlled generation of cationic [LB∙X]+ species is possible by varying the nature of LB, varying the nature of the solvent, and varying the nature of the halogen X. Molecular Cl2 has the greatest tendency to form ionic species in polar solvents. Spontaneous ionization of molecular nσ complexes of chlorine with strong LB in medium-polar solvents (starting from OEt2, ε = 4.24) should not be neglected and single point solvation energy computations on gas phase optimized geometries are not reliable for such systems.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


