{"title":"Molecular profiles and long-term outcomes of Thai children with hepatic glycogen storage disease in Thailand.","authors":"Jaravee Vanduangden, Rungnapa Ittiwut, Chupong Ittiwut, Teerasak Phewplung, Anapat Sanpavat, Palittiya Sintusek, Kanya Suphapeetiporn","doi":"10.5409/wjcp.v13.i4.100493","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Thus far, genetic analysis of patients clinically diagnosed with glycogen storage diseases (GSDs) in Thailand has not been reported.</p><p><strong>Aim: </strong>To evaluate the clinical and biochemical profiles, molecular analysis and long-term outcomes of Thai children diagnosed with hepatic GSD.</p><p><strong>Methods: </strong>Children aged < 18 years diagnosed with hepatic GSD and followed up at King Chulalongkorn Memorial Hospital were recruited. Whole-exome sequencing (WES) was performed to identify the causative gene variants. Medical records were assessed.</p><p><strong>Results: </strong>All eight children with histopathologically confirmed diagnosis were classified by WES into subtypes Ia (<i>n</i> = 1), III (<i>n</i> = 3), VI (<i>n</i> = 3), and IX (<i>n</i> = 1). A total number of 10 variants were identified including <i>G6PC</i> (<i>n</i> = 1), <i>AGL</i> (<i>n</i> = 4), <i>PYGL</i> (<i>n</i> = 5), and <i>PHKA2</i> (<i>n</i> = 1). <i>AGL</i> had two novel variants. The clinical manifestations were hepatomegaly (<i>n</i> = 8), doll-like facies (<i>n</i> = 3), wasting (<i>n</i> = 2), and stunting (<i>n</i> = 5). All patients showed hypoglycemia, transaminitis, and dyslipidemia. The mainstay of treatment was cornstarch supplementation and high-protein and low-lactose-fructose diet. After a median follow-up time of 9.59 years, height turned to normal for age in 3/5 patients and none had malnutrition. Liver enzymes, blood sugar, and lipid profiles improved in all.</p><p><strong>Conclusion: </strong>Hepatomegaly, transaminitis, and hypoglycemia are the hallmarks of GSD confirmed by liver histopathology. Molecular analysis can confirm the diagnosis or classify the subtype that might benefit from personalized treatment, prognosis, and long-term care.</p>","PeriodicalId":75338,"journal":{"name":"World journal of clinical pediatrics","volume":"13 4","pages":"100493"},"PeriodicalIF":0.0000,"publicationDate":"2024-12-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11572614/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"World journal of clinical pediatrics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.5409/wjcp.v13.i4.100493","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
Abstract
Background: Thus far, genetic analysis of patients clinically diagnosed with glycogen storage diseases (GSDs) in Thailand has not been reported.
Aim: To evaluate the clinical and biochemical profiles, molecular analysis and long-term outcomes of Thai children diagnosed with hepatic GSD.
Methods: Children aged < 18 years diagnosed with hepatic GSD and followed up at King Chulalongkorn Memorial Hospital were recruited. Whole-exome sequencing (WES) was performed to identify the causative gene variants. Medical records were assessed.
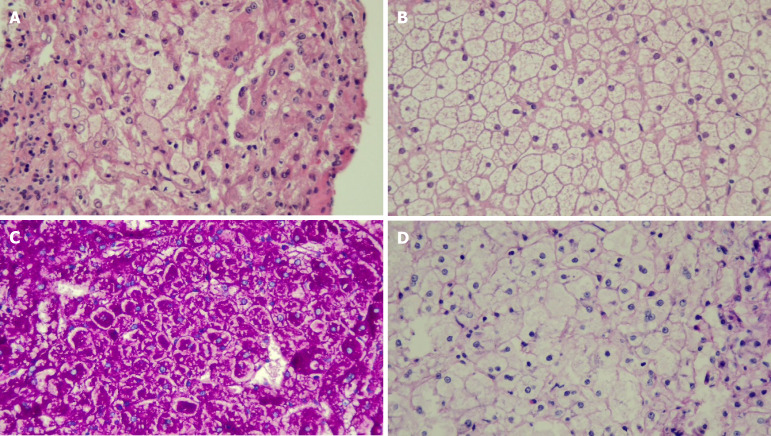
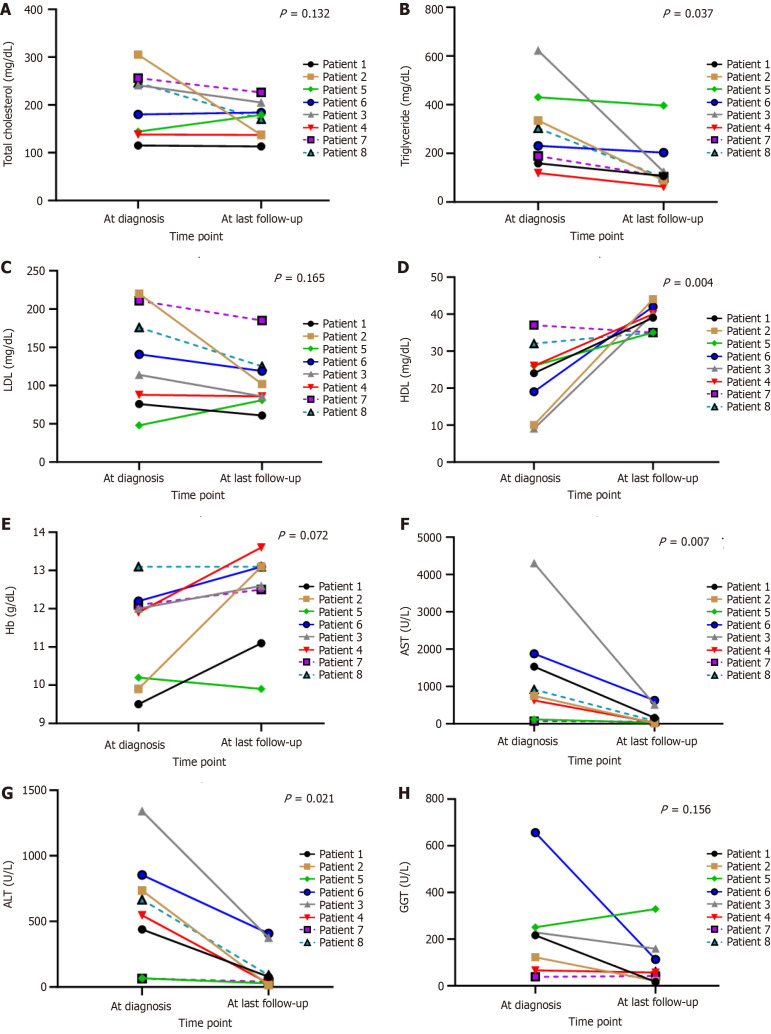
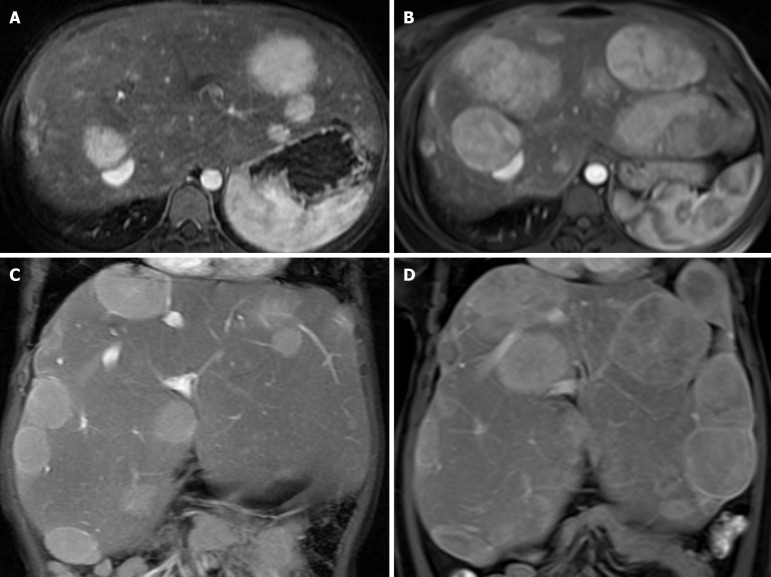
Results: All eight children with histopathologically confirmed diagnosis were classified by WES into subtypes Ia (n = 1), III (n = 3), VI (n = 3), and IX (n = 1). A total number of 10 variants were identified including G6PC (n = 1), AGL (n = 4), PYGL (n = 5), and PHKA2 (n = 1). AGL had two novel variants. The clinical manifestations were hepatomegaly (n = 8), doll-like facies (n = 3), wasting (n = 2), and stunting (n = 5). All patients showed hypoglycemia, transaminitis, and dyslipidemia. The mainstay of treatment was cornstarch supplementation and high-protein and low-lactose-fructose diet. After a median follow-up time of 9.59 years, height turned to normal for age in 3/5 patients and none had malnutrition. Liver enzymes, blood sugar, and lipid profiles improved in all.
Conclusion: Hepatomegaly, transaminitis, and hypoglycemia are the hallmarks of GSD confirmed by liver histopathology. Molecular analysis can confirm the diagnosis or classify the subtype that might benefit from personalized treatment, prognosis, and long-term care.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


