Assessing the Sensitivity of Pourbaix Diagrams to Computational Protocols: Electrochemical Stability of Ni Oxides as a Case Study
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
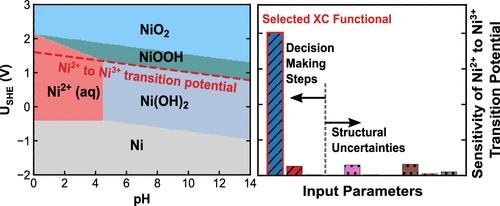
Pourbaix diagrams stand as a useful tool in assessing and visualizing materials’ electrochemical stability and are widely used for electrocatalyst design. However, their reliability hinges on the accuracy of the chemical potentials of involved phases, which may bear uncertainties and can be significantly impacted by decision-making steps in the computational protocol. This study introduces a robust sensitivity analysis framework, exemplified through a detailed examination of the computational Pourbaix diagram of Ni, the oxides of which are used as high-activity and cost-friendly catalysts for many electrochemical reactions. Quantities of interest derived from the Pourbaix diagram include the appearance and stability domain of the catalytically active Ni oxide phases along with the onset electrochemical potentials of phase transitions. These metrics can guide the design of operational conditions for Ni oxide electrocatalysts. We find that the employed DFT exchange-correlation functional has the most significant influence on the computed Pourbaix diagram. Uncertainties on crystal structures, along with their related ab initio energetics, are also found to affect the size of the phase stability domain. Higher-order coupling among input parameters is found to play a crucial role in influencing the appearance and distribution of Ni phases in the diagram. Our findings suggest a need to consider variations and uncertainties associated with the computational procedures on predicted Pourbaix diagrams for materials design.
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


