{"title":"SeqDPI: A 1D-CNN approach for predicting binding affinity of kinase inhibitors","authors":"Vinay Priy Mishra, Yogendra Narain Singh, Feroz Khan, Malay Kishore Dutta","doi":"10.1002/jcc.27518","DOIUrl":null,"url":null,"abstract":"<p>Predicting drug target binding affinity has huge relevance in Modern drug discovery and drug repositioning processes which assist doctors to come up with new drugs or even use the existing drugs for new target proteins. In silico models, using advanced deep learning techniques could further assist these prediction tasks by providing most prominent drug target pairs. Considering these factors, a deep learning based algorithmic framework is developed in this study to support drug target interaction prediction. The proposed <i>SeqDPI</i> model extract the relevant drug and protein features from the one dimensional Sequential representation of the dataset considered using optimized CNN networks that deploy convolutions on varying length of amino acid subsequence's to capture hidden pattern, the convolved drug- protein features obtained are then used as an input to L2 penalized feed forward neural network which matches the local residue patterns in protein classes with molecular fingerprints of drugs to predict the binding strength for all drug target pairs. The proposed model reduces the convolution strain typically encountered in existing in silico models that utilize complex 3D structures of drug protein datasets. The result shows that the SeqDPI model achieves a mean square error MSE of (0.167) across cross validation folds, outperforming baseline models such as KronRLS (0.406), Simboost (0.226), and DeepPS (0.214). Additionally, SeqDPI attains a high CI score of 0.9114 on the benchmark KIBA dataset, demonstrating its statistical significance and computational efficiency compared to existing methods. This gives the relevance and effectiveness of SeqDPI model in accurately predicting binding affinities while working with simpler one-dimensional data, making it a robust and computationally cost-effective solution for drug-target interaction prediction.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-12-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27518","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
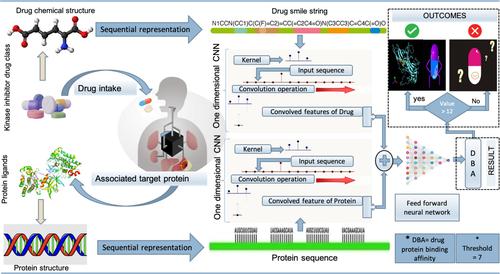
Predicting drug target binding affinity has huge relevance in Modern drug discovery and drug repositioning processes which assist doctors to come up with new drugs or even use the existing drugs for new target proteins. In silico models, using advanced deep learning techniques could further assist these prediction tasks by providing most prominent drug target pairs. Considering these factors, a deep learning based algorithmic framework is developed in this study to support drug target interaction prediction. The proposed SeqDPI model extract the relevant drug and protein features from the one dimensional Sequential representation of the dataset considered using optimized CNN networks that deploy convolutions on varying length of amino acid subsequence's to capture hidden pattern, the convolved drug- protein features obtained are then used as an input to L2 penalized feed forward neural network which matches the local residue patterns in protein classes with molecular fingerprints of drugs to predict the binding strength for all drug target pairs. The proposed model reduces the convolution strain typically encountered in existing in silico models that utilize complex 3D structures of drug protein datasets. The result shows that the SeqDPI model achieves a mean square error MSE of (0.167) across cross validation folds, outperforming baseline models such as KronRLS (0.406), Simboost (0.226), and DeepPS (0.214). Additionally, SeqDPI attains a high CI score of 0.9114 on the benchmark KIBA dataset, demonstrating its statistical significance and computational efficiency compared to existing methods. This gives the relevance and effectiveness of SeqDPI model in accurately predicting binding affinities while working with simpler one-dimensional data, making it a robust and computationally cost-effective solution for drug-target interaction prediction.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


