{"title":"An ab initio study of the rovibronic spectra of CH†","authors":"Zhenlu Hou and Linhua Liu","doi":"10.1039/D4CP03298E","DOIUrl":null,"url":null,"abstract":"<p >CH is one of the most spectroscopically studied diatomic molecules. The rovibronic spectra of the methylidyne radical (CH) in adiabatic and diabatic representations are obtained based on <em>ab initio</em> data, including 12 potential energy curves, 38 dipole moment curves, 79 spin–orbit coupling curves, and 18 electronic angular momentum coupling curves. We employed the internally contracted multireference configuration interaction method including the Davidson correction with the aug-cc-pV(5+d)Z basis set for the C atom and the aug-cc-pV5Z basis set for the H atom. The diabatic transformations are performed based on a property-based diabatisation method to remove the avoided crossings for the E <small><sup>2</sup></small>Π–F <small><sup>2</sup></small>Π and F <small><sup>2</sup></small>Π–H <small><sup>2</sup></small>Π pairs. The coupled nuclear motion Schrödinger equations are then solved using the Duo nuclear motion program to obtain the rovibronic spectra of CH for wavenumbers from 0 to 80 000 cm<small><sup>−1</sup></small> at 5000 K. An overall prediction of the rovibronic spectra of CH is provided in this work. Our results could be beneficial for future calculations of rovibronic spectra of CH and contribute to improving astronomical, chemical, and physical models.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 1","pages":" 367-375"},"PeriodicalIF":2.9000,"publicationDate":"2024-12-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03298e","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
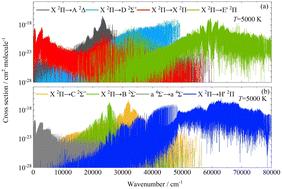
CH is one of the most spectroscopically studied diatomic molecules. The rovibronic spectra of the methylidyne radical (CH) in adiabatic and diabatic representations are obtained based on ab initio data, including 12 potential energy curves, 38 dipole moment curves, 79 spin–orbit coupling curves, and 18 electronic angular momentum coupling curves. We employed the internally contracted multireference configuration interaction method including the Davidson correction with the aug-cc-pV(5+d)Z basis set for the C atom and the aug-cc-pV5Z basis set for the H atom. The diabatic transformations are performed based on a property-based diabatisation method to remove the avoided crossings for the E 2Π–F 2Π and F 2Π–H 2Π pairs. The coupled nuclear motion Schrödinger equations are then solved using the Duo nuclear motion program to obtain the rovibronic spectra of CH for wavenumbers from 0 to 80 000 cm−1 at 5000 K. An overall prediction of the rovibronic spectra of CH is provided in this work. Our results could be beneficial for future calculations of rovibronic spectra of CH and contribute to improving astronomical, chemical, and physical models.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


