Ding Cao, Ekkehard Grünig, Yuriy Sirenko, Ganna Radchenko, Henning Gall, Ayat Ahmed, Susanne Theiß, Mareike Lankeit, Benjamin Meder, Magdalena Laugsch, Christina A Eichstaedt
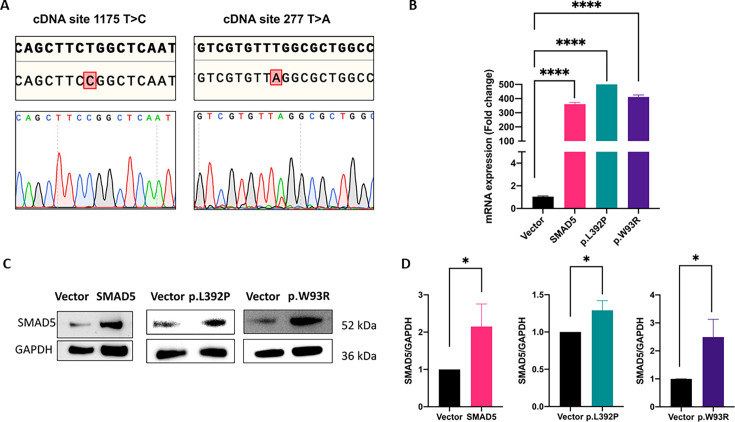
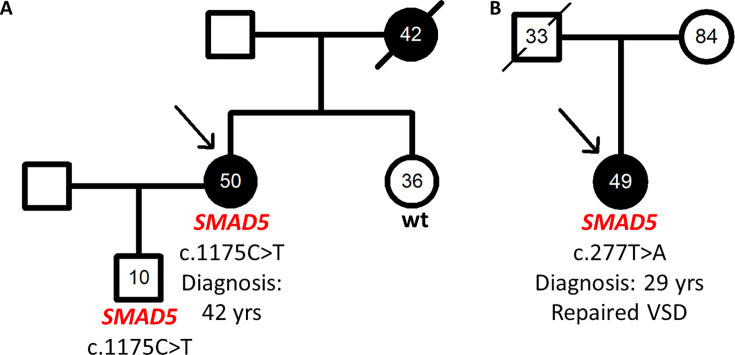
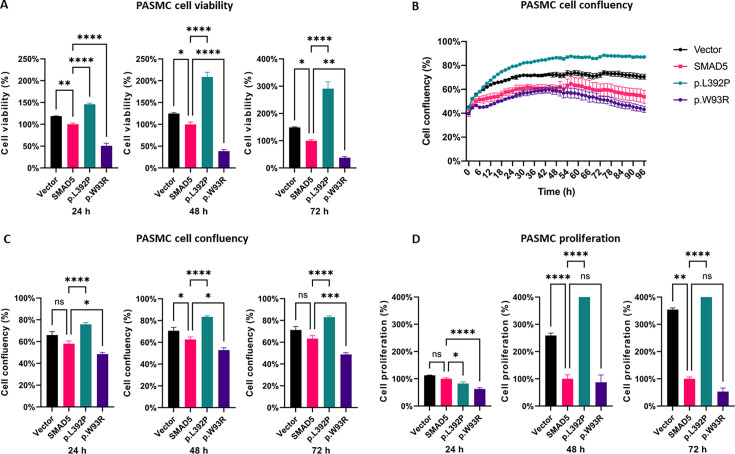
{"title":"SMAD5 as a novel gene for familial pulmonary arterial hypertension.","authors":"Ding Cao, Ekkehard Grünig, Yuriy Sirenko, Ganna Radchenko, Henning Gall, Ayat Ahmed, Susanne Theiß, Mareike Lankeit, Benjamin Meder, Magdalena Laugsch, Christina A Eichstaedt","doi":"10.1042/CS20241340","DOIUrl":null,"url":null,"abstract":"<p><p>Genetic diagnostic testing of 325 pulmonary arterial hypertension (PAH) patients using a PAH specific gene panel including 18 known PAH genes revealed mutations in 23%. Further PAH candidate genes were sequenced in the remaining patients exposing two SMAD5 variants, which were clinically and functionally characterized. We first recorded familial cosegregation and clinical parameters. Functional tests were performed following transient over-expression of the two SMAD5 variants in pulmonary artery smooth muscle cells (PASMCs). Expression of these variants was confirmed by quantitative PCR, Sanger sequencing, and Western blotting. Cell viability was evaluated using cell counting kit 8, cell proliferation by bromodeoxyuridine (BrdU), and apoptosis by annexin V assay. Both SMAD5 missense variants were absent in healthy controls and predicted to be pathogenic. The variant c.1175T>C p.(Leu392Pro) was identified in a heritable PAH patient and her healthy son. The mother had died of suspected PAH at age 42. The expression of this variant in PASMCs led to significantly higher cell viability due to higher proliferation in comparison with SMAD5 wild-type cells. The second variant c.277T>A p.(Trp93Arg) was identified in a patient with congenital heart disease associated PAH with a surgically repaired ventricular septal defect. Its expression led to significantly lower cell viability due to increased apoptosis in comparison with wild-type SMAD5 cells. Taking into account familial aggregation, clinical findings, and functional evidence, both variants could be classified as likely pathogenic. This is the first description of SMAD5 as a potential novel PAH gene for genetic diagnostic testing.</p>","PeriodicalId":10475,"journal":{"name":"Clinical science","volume":" ","pages":"15-27"},"PeriodicalIF":7.7000,"publicationDate":"2025-01-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC12204011/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical science","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1042/CS20241340","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
Genetic diagnostic testing of 325 pulmonary arterial hypertension (PAH) patients using a PAH specific gene panel including 18 known PAH genes revealed mutations in 23%. Further PAH candidate genes were sequenced in the remaining patients exposing two SMAD5 variants, which were clinically and functionally characterized. We first recorded familial cosegregation and clinical parameters. Functional tests were performed following transient over-expression of the two SMAD5 variants in pulmonary artery smooth muscle cells (PASMCs). Expression of these variants was confirmed by quantitative PCR, Sanger sequencing, and Western blotting. Cell viability was evaluated using cell counting kit 8, cell proliferation by bromodeoxyuridine (BrdU), and apoptosis by annexin V assay. Both SMAD5 missense variants were absent in healthy controls and predicted to be pathogenic. The variant c.1175T>C p.(Leu392Pro) was identified in a heritable PAH patient and her healthy son. The mother had died of suspected PAH at age 42. The expression of this variant in PASMCs led to significantly higher cell viability due to higher proliferation in comparison with SMAD5 wild-type cells. The second variant c.277T>A p.(Trp93Arg) was identified in a patient with congenital heart disease associated PAH with a surgically repaired ventricular septal defect. Its expression led to significantly lower cell viability due to increased apoptosis in comparison with wild-type SMAD5 cells. Taking into account familial aggregation, clinical findings, and functional evidence, both variants could be classified as likely pathogenic. This is the first description of SMAD5 as a potential novel PAH gene for genetic diagnostic testing.
期刊介绍:
Translating molecular bioscience and experimental research into medical insights, Clinical Science offers multi-disciplinary coverage and clinical perspectives to advance human health.
Its international Editorial Board is charged with selecting peer-reviewed original papers of the highest scientific merit covering the broad spectrum of biomedical specialities including, although not exclusively:
Cardiovascular system
Cerebrovascular system
Gastrointestinal tract and liver
Genomic medicine
Infection and immunity
Inflammation
Oncology
Metabolism
Endocrinology and nutrition
Nephrology
Circulation
Respiratory system
Vascular biology
Molecular pathology.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


