Assessment of DFT Functionals for Predicting the Magnetic Exchange Coupling Constants of Nonalternant Hydrocarbon Diradicals: The Role of Hartree–Fock Exchange
{"title":"Assessment of DFT Functionals for Predicting the Magnetic Exchange Coupling Constants of Nonalternant Hydrocarbon Diradicals: The Role of Hartree–Fock Exchange","authors":"Suranjan Shil","doi":"10.1002/jcc.27531","DOIUrl":null,"url":null,"abstract":"<p>The magnetic nature of nonalternant hydrocarbon (Azulene) bridged nitronyl nitroxide (AzNN<sub>2</sub>) and imino-nitroxide (AzIN<sub>2</sub>) diradicals are investigated with 38 different DFT functionals to find out a correct functional to predict the magnetic nature of these diradicals. The effect of Hartree–Fock exchange (HFX) in the hybrid functionals are investigated for the prediction of magnetic nature of the nonalternant hydrocarbon bridged diradicals. The utility of Borden and Davidson's proposal of disjoint and nondisjoint SOMOs for the prediction of magnetic nature of alternant hydrocarbon bridged diradicals is assessed for the nonalternant hydrocarbon based diradicals. The more affordable meta-GGA functionals was found to be outperforming the costlier hybrid and double-hybrid functionals in predicting the magnetic properties of nonalternant hydrocarbon-bridged diradicals. HFX significantly influences a functional's ability to predict a diradical's magnetic nature. Interestingly, Borden and Davidson's concept of disjoint and nondisjoint SOMOs, which is used to predict the magnetic behavior of alternant hydrocarbon diradicals, is reversed for nonalternant hydrocarbon-bridged diradicals. The difference in the magnetic nature of the two diradicals come from the canonical molecular orbitals of the diradicals, one has set of disjoint SOMOs and other has nondisjoint SOMOs.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27531","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27531","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
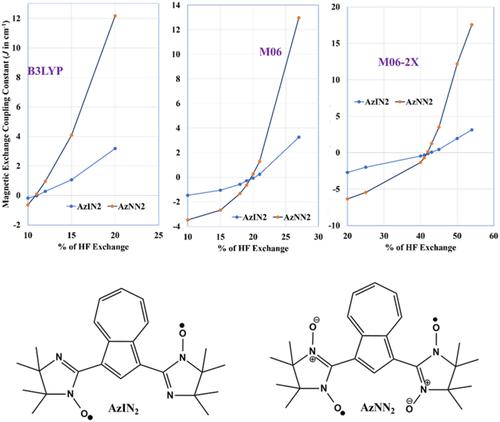
The magnetic nature of nonalternant hydrocarbon (Azulene) bridged nitronyl nitroxide (AzNN2) and imino-nitroxide (AzIN2) diradicals are investigated with 38 different DFT functionals to find out a correct functional to predict the magnetic nature of these diradicals. The effect of Hartree–Fock exchange (HFX) in the hybrid functionals are investigated for the prediction of magnetic nature of the nonalternant hydrocarbon bridged diradicals. The utility of Borden and Davidson's proposal of disjoint and nondisjoint SOMOs for the prediction of magnetic nature of alternant hydrocarbon bridged diradicals is assessed for the nonalternant hydrocarbon based diradicals. The more affordable meta-GGA functionals was found to be outperforming the costlier hybrid and double-hybrid functionals in predicting the magnetic properties of nonalternant hydrocarbon-bridged diradicals. HFX significantly influences a functional's ability to predict a diradical's magnetic nature. Interestingly, Borden and Davidson's concept of disjoint and nondisjoint SOMOs, which is used to predict the magnetic behavior of alternant hydrocarbon diradicals, is reversed for nonalternant hydrocarbon-bridged diradicals. The difference in the magnetic nature of the two diradicals come from the canonical molecular orbitals of the diradicals, one has set of disjoint SOMOs and other has nondisjoint SOMOs.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


