A First-Principle Study Investigating the Half-Metallic and Mechanical Properties of Double Halide Perovskites Rb2OsX6 (X = cl, Br, and I) for Spintronic Applications
{"title":"A First-Principle Study Investigating the Half-Metallic and Mechanical Properties of Double Halide Perovskites Rb2OsX6 (X = cl, Br, and I) for Spintronic Applications","authors":"Mohamed Boubchir, Zeyneb Bordjiba, Rabie Amraoui, Rachid Boubchir, Hafid Aourag","doi":"10.1002/jcc.27537","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>In this work, we present a density functional calculation of the structural, electronic, and mechanical properties of cubic double halide perovskites Rb<sub>2</sub>OsX<sub>6</sub> (X = Cl, Br, and I). Our results show that these compounds are stable in the ferromagnetic phase with lattice parameters, bulk modulus, and their first-pressure derivatives in good agreement with other available theoretical data. The negative values of cohesive energy and formation energy, along with the absence of negative or imaginary frequencies in the phonon spectrum, confirm the mechanical stability of all the compounds. The Curie temperature (Tc) is determined using a Heisenberg model in the mean-field approximation. We obtained a half-metallic character for all compounds, making them promising materials for spintronic applications. The magnetic properties indicate that the Os atoms in all compounds are responsible for the magnetism, while the positive exchange constants suggest a strong preference for ferromagnetic alignment. This indicates a stable ferromagnetic phase and potential applications in spintronics. The mechanical properties demonstrate that the compounds studied are isotropic and ductile.</p>\n </div>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27537","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
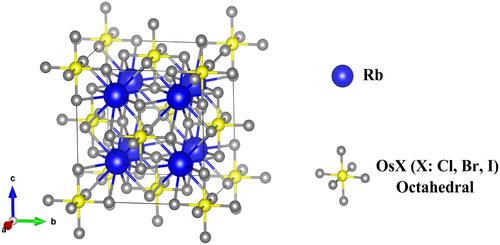
In this work, we present a density functional calculation of the structural, electronic, and mechanical properties of cubic double halide perovskites Rb2OsX6 (X = Cl, Br, and I). Our results show that these compounds are stable in the ferromagnetic phase with lattice parameters, bulk modulus, and their first-pressure derivatives in good agreement with other available theoretical data. The negative values of cohesive energy and formation energy, along with the absence of negative or imaginary frequencies in the phonon spectrum, confirm the mechanical stability of all the compounds. The Curie temperature (Tc) is determined using a Heisenberg model in the mean-field approximation. We obtained a half-metallic character for all compounds, making them promising materials for spintronic applications. The magnetic properties indicate that the Os atoms in all compounds are responsible for the magnetism, while the positive exchange constants suggest a strong preference for ferromagnetic alignment. This indicates a stable ferromagnetic phase and potential applications in spintronics. The mechanical properties demonstrate that the compounds studied are isotropic and ductile.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


