Density Functional Theory (DFT) and Time-Dependent DFT (TDDFT) Studies of Porphyrin Adsorption on Graphene: Insights on the Effect of Substituents and Central Metal on Adsorption Energies
Rayene Gara, Ángel Morales-García, Youssef Arfaoui, Francesc Illas
{"title":"Density Functional Theory (DFT) and Time-Dependent DFT (TDDFT) Studies of Porphyrin Adsorption on Graphene: Insights on the Effect of Substituents and Central Metal on Adsorption Energies","authors":"Rayene Gara, Ángel Morales-García, Youssef Arfaoui, Francesc Illas","doi":"10.1002/jcc.27526","DOIUrl":null,"url":null,"abstract":"<p>Combining metalloporphyrins (MPr) and graphene constitutes key composites in the development of photovoltaic devices. Here, we focus on the analysis of the properties of metalloporphyrins/graphene systems by means of the density functional theory (DFT) and its time-dependent (TDDFT) version, focusing on the ground and singlet excited states. Our benchmark analysis concludes that ωB97XD density functional combined with 6-31G(d)/Def2-TZVP basis set is a better-suited method for simulating accurate MPr adsorption on graphene. It is shown that a reduced atomic model where the external organic shell of the structure is removed provides the same resulting optoelectronic properties of the original model, constituting an important speed-up of the calculations when studying porphyrins-derived molecules. We observe that ZnPr provides the highest light harvesting efficiency (LHE) value. In addition, we find out that the adsorption energy increases monotonically with the size of the graphene flake and the highest stability involves the use of graphene comprising above 500 atoms. Besides, CdPr and HgPr keep their properties as photosensitizers when they are bonded to graphene and show promising values in terms of LHE emerging as suitable solar energy harvesters.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-12-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27526","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27526","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
引用次数: 0
Abstract
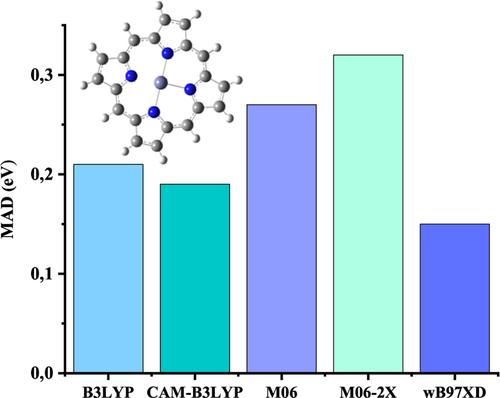
Combining metalloporphyrins (MPr) and graphene constitutes key composites in the development of photovoltaic devices. Here, we focus on the analysis of the properties of metalloporphyrins/graphene systems by means of the density functional theory (DFT) and its time-dependent (TDDFT) version, focusing on the ground and singlet excited states. Our benchmark analysis concludes that ωB97XD density functional combined with 6-31G(d)/Def2-TZVP basis set is a better-suited method for simulating accurate MPr adsorption on graphene. It is shown that a reduced atomic model where the external organic shell of the structure is removed provides the same resulting optoelectronic properties of the original model, constituting an important speed-up of the calculations when studying porphyrins-derived molecules. We observe that ZnPr provides the highest light harvesting efficiency (LHE) value. In addition, we find out that the adsorption energy increases monotonically with the size of the graphene flake and the highest stability involves the use of graphene comprising above 500 atoms. Besides, CdPr and HgPr keep their properties as photosensitizers when they are bonded to graphene and show promising values in terms of LHE emerging as suitable solar energy harvesters.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.


 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


