Theoretical insights into the effect of metal co-substituted CeO2(111) surfaces on oxygen vacancy formation and chemical looping CO2 assisted CH4 conversion to synthesis gas†
Zeshan Wang, Yuelun Li, Yuxin Wang, Tao Li, Jiahao Zheng, LiNan Huang, Huicong Zuo, Dong Tian, Hua Wang and Kongzhai Li
{"title":"Theoretical insights into the effect of metal co-substituted CeO2(111) surfaces on oxygen vacancy formation and chemical looping CO2 assisted CH4 conversion to synthesis gas†","authors":"Zeshan Wang, Yuelun Li, Yuxin Wang, Tao Li, Jiahao Zheng, LiNan Huang, Huicong Zuo, Dong Tian, Hua Wang and Kongzhai Li","doi":"10.1039/D4CP03370A","DOIUrl":null,"url":null,"abstract":"<p >The density functional theory (DFT) method is used to investigate the effect of low oxygen vacancy formation energy on the catalytic performance of chemical looping dry reforming of methane (CL-DRM) when metal ions are co-substituted on CeO<small><sub>2</sub></small>(111) surfaces. The results show that the oxygen vacancy formation energy is extremely low with a value of −2.05 eV when Zn and Nd are co-substituted on the CeO<small><sub>2</sub></small>(111) surface. For the CH<small><sub>4</sub></small> conversion process in CL-DRM, the reaction paths are found to be CH<small><sub>4</sub></small> → CH<small><sub>3</sub></small> → CH<small><sub>2</sub></small> → CH → C → CO paths on the pristine as well as on the Zn and Nd co-substituted surfaces. The critical rate-limiting step for both pristine and co-substituted surfaces is the dehydrogenation of CH<small><sub>2</sub></small> to form CH and H with activation energies of 1.62 and 1.00 eV, respectively. This indicates that the surface co-substituted with Zn and Nd promotes the CH<small><sub>4</sub></small> conversion process more effectively than the clean surface. However, the desorption process of syngas on the co-substituted surface requires high energy, and CO is easily peroxidized to CO<small><sub>2</sub></small> before desorption, reducing the selectivity of CO to the detriment of syngas production. For the CO<small><sub>2</sub></small> cleavage process in CL-DRM, it is difficult for CO<small><sub>2</sub></small> to generate enough energy on the co-substituted surfaces to overcome the activation energy of the reaction. The formation of oxygen vacancies is facilitated by an extremely low oxygen vacancy formation energy, which in turn enhances the adsorption of reaction intermediates in the CL-DRM process onto the oxygen carrier. Nevertheless, an excessive accumulation of oxygen vacancies can drive the oxygen carrier into a hyperactivated condition, which may inhibit the desired reaction pathways and reduce the efficiency and selectivity of the CL-DRM process. The present study is of great importance for the design concept of oxygen carriers in CL-DRM and the application potential of oxygen vacancy regulation.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 2","pages":" 868-884"},"PeriodicalIF":2.9000,"publicationDate":"2024-12-02","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2025/cp/d4cp03370a","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
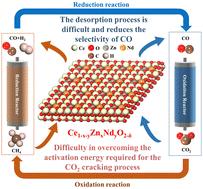
The density functional theory (DFT) method is used to investigate the effect of low oxygen vacancy formation energy on the catalytic performance of chemical looping dry reforming of methane (CL-DRM) when metal ions are co-substituted on CeO2(111) surfaces. The results show that the oxygen vacancy formation energy is extremely low with a value of −2.05 eV when Zn and Nd are co-substituted on the CeO2(111) surface. For the CH4 conversion process in CL-DRM, the reaction paths are found to be CH4 → CH3 → CH2 → CH → C → CO paths on the pristine as well as on the Zn and Nd co-substituted surfaces. The critical rate-limiting step for both pristine and co-substituted surfaces is the dehydrogenation of CH2 to form CH and H with activation energies of 1.62 and 1.00 eV, respectively. This indicates that the surface co-substituted with Zn and Nd promotes the CH4 conversion process more effectively than the clean surface. However, the desorption process of syngas on the co-substituted surface requires high energy, and CO is easily peroxidized to CO2 before desorption, reducing the selectivity of CO to the detriment of syngas production. For the CO2 cleavage process in CL-DRM, it is difficult for CO2 to generate enough energy on the co-substituted surfaces to overcome the activation energy of the reaction. The formation of oxygen vacancies is facilitated by an extremely low oxygen vacancy formation energy, which in turn enhances the adsorption of reaction intermediates in the CL-DRM process onto the oxygen carrier. Nevertheless, an excessive accumulation of oxygen vacancies can drive the oxygen carrier into a hyperactivated condition, which may inhibit the desired reaction pathways and reduce the efficiency and selectivity of the CL-DRM process. The present study is of great importance for the design concept of oxygen carriers in CL-DRM and the application potential of oxygen vacancy regulation.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


