{"title":"Theoretical study on the thermal decomposition mechanism of 2-nitro-[1,2,4]triazolo[1,5-a][1,3,5]triazine-5,7-diamine","authors":"Mengjie Bo, Jun Cao, Congming Ma, Peng Ma","doi":"10.1007/s00894-024-06228-0","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>High nitrogen and high-density compounds have become popular research objects in the energetic materials in recent years. Among them, compounds composed of triazine and azole skeleton rings have received attention due to their good stability and nitrogen content. The triazine imidazole-based condensed ring energetic derivatives have good properties and lack research on the direction of thermal decomposition. By exploring the thermal decomposition mechanism of energetic materials, their safety and thermal stability can be effectively analyzed. 2-Nitro-[1,2,4]triazolo[1,5-a][1,3,5]triazine-5,7-diamine has excellent thermal stability and low mechanical sensitivity, making it a potential candidate for heat-resistant and insensitive energetic materials. So this article chooses 2-nitro-[1,2,4] triazolo[1,5-a][1,3,5]triazine-5,7-diamine as the research object and uses density functional theory (DFT) to study its thermal decomposition mechanism. The thermal decomposition mechanism is helpful to deepen the understanding of these substances. This work calculated the key information such as the reaction potential barrier of the substance and gradually derived it. The research shows that its decomposition path includes ring cleavage, hydrogen atom rearrangement, and free radical detachment.</p><h3>Methods</h3><p>Based on the density functional theory (DFT), all substances in this work were subjected to structural optimization and energy calculations using the B3LYP/6–311 + G(d,p) and M06-2X/def2TZVPP methods. After optimizing convergence, perform vibration analysis without imaginary frequencies to obtain a stable structure.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 12","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-11-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06228-0","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
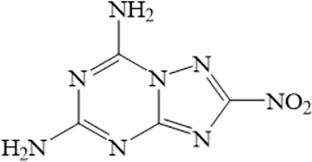
High nitrogen and high-density compounds have become popular research objects in the energetic materials in recent years. Among them, compounds composed of triazine and azole skeleton rings have received attention due to their good stability and nitrogen content. The triazine imidazole-based condensed ring energetic derivatives have good properties and lack research on the direction of thermal decomposition. By exploring the thermal decomposition mechanism of energetic materials, their safety and thermal stability can be effectively analyzed. 2-Nitro-[1,2,4]triazolo[1,5-a][1,3,5]triazine-5,7-diamine has excellent thermal stability and low mechanical sensitivity, making it a potential candidate for heat-resistant and insensitive energetic materials. So this article chooses 2-nitro-[1,2,4] triazolo[1,5-a][1,3,5]triazine-5,7-diamine as the research object and uses density functional theory (DFT) to study its thermal decomposition mechanism. The thermal decomposition mechanism is helpful to deepen the understanding of these substances. This work calculated the key information such as the reaction potential barrier of the substance and gradually derived it. The research shows that its decomposition path includes ring cleavage, hydrogen atom rearrangement, and free radical detachment.
Methods
Based on the density functional theory (DFT), all substances in this work were subjected to structural optimization and energy calculations using the B3LYP/6–311 + G(d,p) and M06-2X/def2TZVPP methods. After optimizing convergence, perform vibration analysis without imaginary frequencies to obtain a stable structure.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


