Insights on formation of oxide layers, corrosion, and hydrogen embrittlement on the Ti2AlNb (1 1 0) surface: Density functional theory study
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
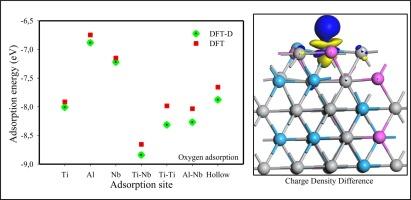
Ti2AlNb alloys offer good mechanical qualities and promise for use in various applications, such as aero-engines and other industries. However, corrosion and hydrogen embrittlement remain important concerns and limitations for their use. In this work, first-principle density functional theory is used to investigate the adsorption of hydrogen, fluorine and oxygen on the surface of Ti2AlNb (1 1 0). The effects of the considered adsorbates on the surface were compared by analysing the adsorption energy, charge density differences, density of states, and work function. The current findings revealed that the adsorption behaviour of all the adsorbates is exothermic and spontaneous due to the negative adsorption energy. More importantly, the effect of Van der Waals forces and dispersion correction was considered, it was found that for all adsorbates the dispersion correction approach exhibited the most stable adsorption energies () than the standard density functional theory (). Thus, the standard DFT underestimates the adsorption energy. Furthermore, it was shown that the adsorption energy strength is dependent on the surface adsorption site, with the Ti-Nb and Al-Nb bridge sites being the most preferred sites for hydrogen, fluorine and oxygen adsorption. Subsequently, it was discovered that oxygen adsorption on the surface of Ti2AlNb (1 1 0) was more thermodynamically stable than hydrogen and fluorine. This suggests that the Ti2AlNb surface will likely suffer from oxidation rather than corrosion and hydrogen embrittlement. In addition, surface atoms showed electron-charge depletion, while adsorbates showed charge accumulation. The adsorption caused charge density redistribution and altered the surface work function.
Ti2AlNb(110)表面氧化层、腐蚀和氢脆的形成:密度泛函理论研究
Ti2AlNb合金具有良好的机械质量,有望用于各种应用,如航空发动机和其他行业。然而,腐蚀和氢脆仍然是其使用的重要问题和限制。本文采用第一性原理密度泛函理论研究了氢、氟和氧在Ti2AlNb(11 10)表面的吸附,通过分析吸附能、电荷密度差、态密度和功函数,比较了不同吸附剂对表面的吸附效果。目前的研究结果表明,由于吸附能为负,所有吸附剂的吸附行为都是放热自发的。更重要的是,考虑了范德华力和色散校正的影响,发现对于所有吸附物,色散校正方法比标准密度泛函理论(EadsDFT)表现出最稳定的吸附能(EadsDFT- d)。因此,标准DFT低估了吸附能。此外,吸附能强度与表面吸附位有关,Ti-Nb和Al-Nb桥位是氢、氟和氧的首选吸附位。随后,我们发现氧在Ti2AlNb(11 10)表面的吸附比氢和氟更具有热力学稳定性。这表明Ti2AlNb表面可能遭受氧化而不是腐蚀和氢脆。此外,表面原子表现为电子电荷耗尽,而吸附剂表现为电荷积累。吸附引起电荷密度重分布,改变了表面功函数。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


