Geometrical features, stability and hydrogen positions in (Al2Cu)n clusters
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
The phase equilibria of the Al-Cu alloys have been well-established, with the Al2Cu being a crucial component of the phase diagram. Constructing an atomic model with a 2:1 stoichiometric ratio of Al and Cu holds significance for further investigating the local structures of the alloy phases. Here, we employ the GA-DFT method to explore the structural potential energy surfaces of (Al2Cu)n clusters (n = 1–6). The results reveal that the (Al2Cu)n evolve from hollow cages to more densely packed configurations, with Al atoms relatively more concentrated and Cu atoms becoming more dispersed throughout the structures. The Eb and Δ2E analyses show that the (Al2Cu)3 has a higher stability than that of its neighbors, and the AIMD simulations demonstrate that it can maintain the structural integrity at 700 K. The molecular orbitals reveal that 21 valence electrons of the (Al2Cu)3 fill superatomic orbits resulting in an electronic configuration of 1S21P61D102S21F1, which is also confirmed by the density of states. The good stability of the (Al2Cu)3 allows the bonding of the H atom to it without causing significant deformation changes in the parent geometry, in which the H tends to preferentially locate at Al sites. The deformation of structures is particularly obvious when H is close to Cu atom.
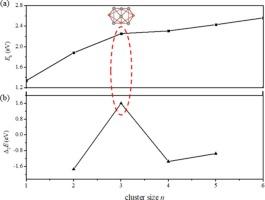
(Al2Cu)n簇的几何特征、稳定性和氢的位置
Al-Cu合金的相平衡已经建立,其中Al2Cu是相图的重要组成部分。建立Al和Cu化学计量比为2:1的原子模型,对进一步研究合金相的局部结构具有重要意义。本文采用GA-DFT方法研究了(Al2Cu)n簇(n = 1-6)的结构势能面。结果表明,(Al2Cu)n从中空笼形结构演变为更密集的排列构型,其中Al原子相对更集中,Cu原子在整个结构中变得更分散。Eb和Δ2E分析表明,(Al2Cu)3具有更高的稳定性,AIMD模拟表明,它在700 K时可以保持结构的完整性。分子轨道表明,(Al2Cu)3的21个价电子填充了超原子轨道,导致电子构型为1S21P61D102S21F1,这也被态密度所证实。(Al2Cu)3良好的稳定性使得H原子与之结合不会引起母体几何结构的明显变形变化,其中H倾向于优先定位于Al位点。当H靠近Cu原子时,结构的变形尤为明显。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


