Denish Trivedi, Kalyani Patrikar and Anirban Mondal
{"title":"Graph-based networks for accurate prediction of ground and excited state molecular properties from minimal features†","authors":"Denish Trivedi, Kalyani Patrikar and Anirban Mondal","doi":"10.1039/D4ME00113C","DOIUrl":null,"url":null,"abstract":"<p >Graph neural networks (GNN) have been demonstrated to correlate molecular structure with properties, enabling rapid evaluation of molecules for a given application. Molecular properties, including ground and excited states, are crucial to analyzing molecular behavior. However, while attention-based mechanisms and pooling methods have been optimized to accurately predict specific properties, no versatile models can predict diverse molecular properties. Here, we present graph neural networks that predict a wide range of properties with high accuracy. Model performance is high regardless of dataset size and origin. Further, we demonstrate an implementation of hierarchical pooling enabling high-accuracy prediction of excited state properties by effectively weighing aspects of features that correlate better with target properties. We show that graph attention networks consistently outperform convolution networks and linear regression, particularly for small dataset sizes. The graph attention model is more accurate than previous message-passing neural networks developed for the prediction of diverse molecular properties. Hence, the model is an efficient tool for screening and designing molecules for applications that require tuning multiple molecular properties.</p>","PeriodicalId":91,"journal":{"name":"Molecular Systems Design & Engineering","volume":" 12","pages":" 1275-1284"},"PeriodicalIF":3.2000,"publicationDate":"2024-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Systems Design & Engineering","FirstCategoryId":"5","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/me/d4me00113c","RegionNum":3,"RegionCategory":"工程技术","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
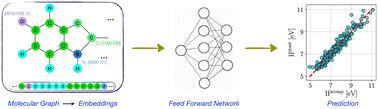
Graph neural networks (GNN) have been demonstrated to correlate molecular structure with properties, enabling rapid evaluation of molecules for a given application. Molecular properties, including ground and excited states, are crucial to analyzing molecular behavior. However, while attention-based mechanisms and pooling methods have been optimized to accurately predict specific properties, no versatile models can predict diverse molecular properties. Here, we present graph neural networks that predict a wide range of properties with high accuracy. Model performance is high regardless of dataset size and origin. Further, we demonstrate an implementation of hierarchical pooling enabling high-accuracy prediction of excited state properties by effectively weighing aspects of features that correlate better with target properties. We show that graph attention networks consistently outperform convolution networks and linear regression, particularly for small dataset sizes. The graph attention model is more accurate than previous message-passing neural networks developed for the prediction of diverse molecular properties. Hence, the model is an efficient tool for screening and designing molecules for applications that require tuning multiple molecular properties.
期刊介绍:
Molecular Systems Design & Engineering provides a hub for cutting-edge research into how understanding of molecular properties, behaviour and interactions can be used to design and assemble better materials, systems, and processes to achieve specific functions. These may have applications of technological significance and help address global challenges.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


