Ivo Fierro-Monti, Klemens Fröhlich, Christian Schori, Alexander Schmidt
{"title":"Assessment of Data-Independent Acquisition Mass Spectrometry (DIA-MS) for the Identification of Single Amino Acid Variants.","authors":"Ivo Fierro-Monti, Klemens Fröhlich, Christian Schori, Alexander Schmidt","doi":"10.3390/proteomes12040033","DOIUrl":null,"url":null,"abstract":"<p><p>Proteogenomics integrates genomic and proteomic data to elucidate cellular processes by identifying variant peptides, including single amino acid variants (SAAVs). In this study, we assessed the capability of data-independent acquisition mass spectrometry (DIA-MS) to identify SAAV peptides in HeLa cells using various search engine pipelines. We developed a customised sequence database (DB) incorporating SAAV sequences from the HeLa genome and conducted searches using DIA-NN, Spectronaut, and Fragpipe-MSFragger. Our evaluation focused on identifying true positive SAAV peptides and false positives through entrapment DBs. This study revealed that DIA-MS provides reproducible and comprehensive coverage of the proteome, identifying a substantial proportion of SAAV peptides. Notably, the DIA-MS searches maintained consistent identification of SAAV peptides despite varying sizes of the entrapment DB. A comparative analysis showed that Fragpipe-MSFragger (FP-DIA) demonstrated the most conservative and effective performance, exhibiting the lowest false discovery match ratio (FDMR). Additionally, integrating DIA and data-dependent acquisition (DDA) MS data search outputs enhanced SAAV peptide identification, with a lower false discovery rate (FDR) observed in DDA searches. The validation using stable isotope dilution and parallel reaction monitoring (SID-PRM) confirmed the SAAV peptides identified by DIA-MS and DDA-MS searches, highlighting the reliability of our approach. Our findings underscore the effectiveness of DIA-MS in proteogenomic workflows for identifying SAAV peptides, offering insights into optimising search engine pipelines and DB construction for accurate proteomics analysis. These methodologies advance the understanding of proteome variability, contributing to cancer research and the identification of novel proteoform therapeutic targets.</p>","PeriodicalId":20877,"journal":{"name":"Proteomes","volume":"12 4","pages":""},"PeriodicalIF":3.6000,"publicationDate":"2024-11-06","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11587465/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Proteomes","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3390/proteomes12040033","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
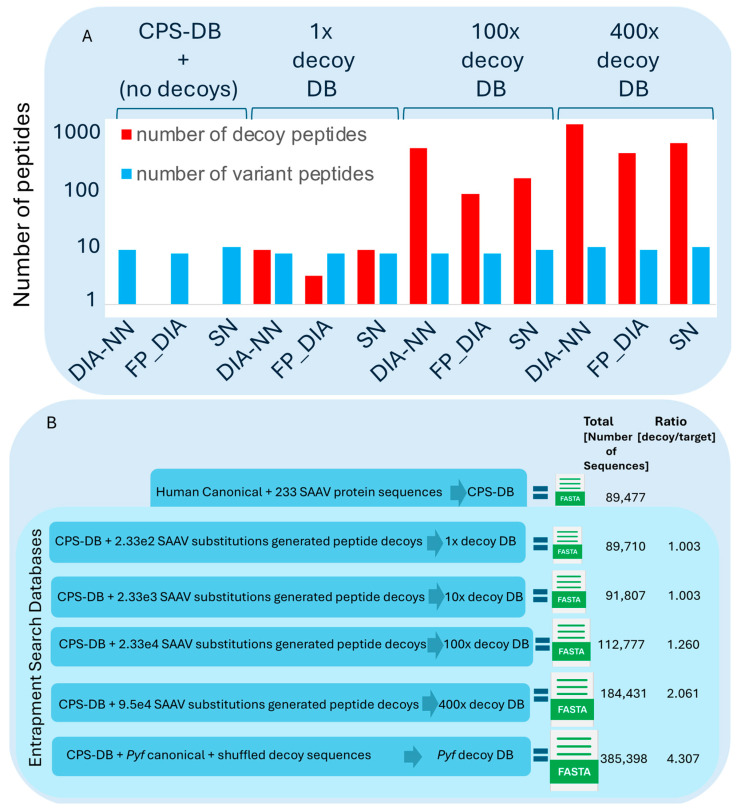
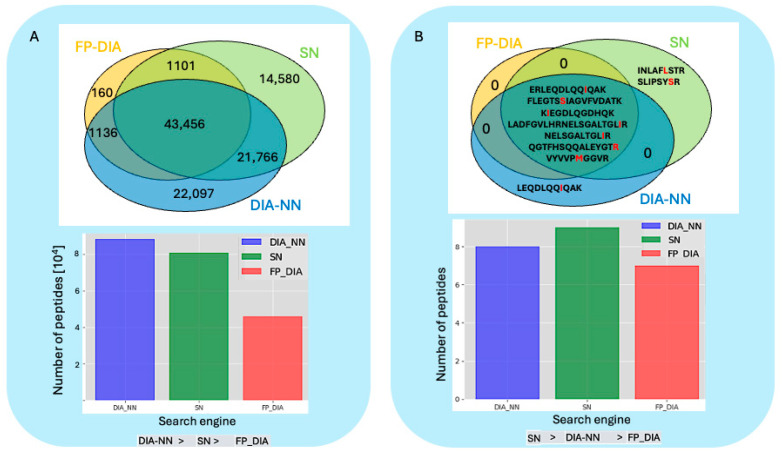
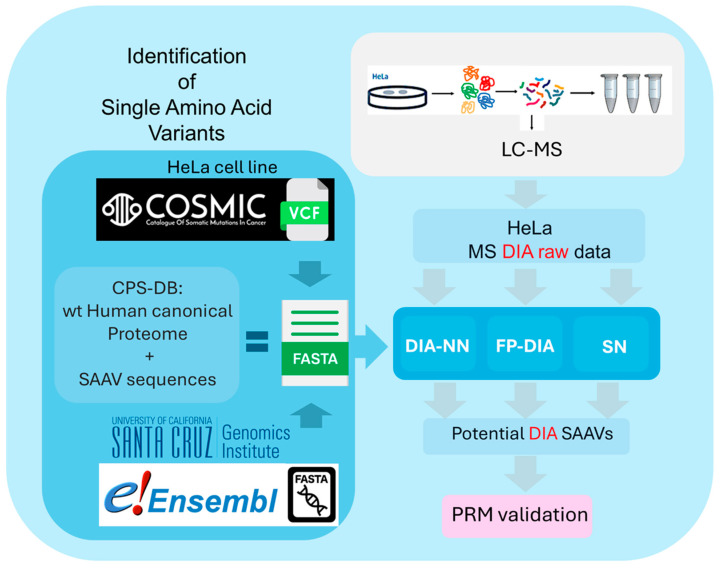
Proteogenomics integrates genomic and proteomic data to elucidate cellular processes by identifying variant peptides, including single amino acid variants (SAAVs). In this study, we assessed the capability of data-independent acquisition mass spectrometry (DIA-MS) to identify SAAV peptides in HeLa cells using various search engine pipelines. We developed a customised sequence database (DB) incorporating SAAV sequences from the HeLa genome and conducted searches using DIA-NN, Spectronaut, and Fragpipe-MSFragger. Our evaluation focused on identifying true positive SAAV peptides and false positives through entrapment DBs. This study revealed that DIA-MS provides reproducible and comprehensive coverage of the proteome, identifying a substantial proportion of SAAV peptides. Notably, the DIA-MS searches maintained consistent identification of SAAV peptides despite varying sizes of the entrapment DB. A comparative analysis showed that Fragpipe-MSFragger (FP-DIA) demonstrated the most conservative and effective performance, exhibiting the lowest false discovery match ratio (FDMR). Additionally, integrating DIA and data-dependent acquisition (DDA) MS data search outputs enhanced SAAV peptide identification, with a lower false discovery rate (FDR) observed in DDA searches. The validation using stable isotope dilution and parallel reaction monitoring (SID-PRM) confirmed the SAAV peptides identified by DIA-MS and DDA-MS searches, highlighting the reliability of our approach. Our findings underscore the effectiveness of DIA-MS in proteogenomic workflows for identifying SAAV peptides, offering insights into optimising search engine pipelines and DB construction for accurate proteomics analysis. These methodologies advance the understanding of proteome variability, contributing to cancer research and the identification of novel proteoform therapeutic targets.
ProteomesBiochemistry, Genetics and Molecular Biology-Clinical Biochemistry
CiteScore
6.50
自引率
3.00%
发文量
37
审稿时长
11 weeks
期刊介绍:
Proteomes (ISSN 2227-7382) is an open access, peer reviewed journal on all aspects of proteome science. Proteomes covers the multi-disciplinary topics of structural and functional biology, protein chemistry, cell biology, methodology used for protein analysis, including mass spectrometry, protein arrays, bioinformatics, HTS assays, etc. Our aim is to encourage scientists to publish their experimental and theoretical results in as much detail as possible. Therefore, there is no restriction on the length of papers. Scope: -whole proteome analysis of any organism -disease/pharmaceutical studies -comparative proteomics -protein-ligand/protein interactions -structure/functional proteomics -gene expression -methodology -bioinformatics -applications of proteomics

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


