Keto-enol Tautomerism of hydroxy-substituted arenes: Theoretical study and experimental consequences
IF 2.7
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
In this work, the chemical equilibrium between enol and keto tautomers occurring in phenol, naphthols and selected 29 hydroxy substituted polycyclic aromatic hydrocarbons classified into 4 structural types was investigated. The reaction Gibbs energies were computed using the density functional theory combined with the solvent continuum model. We have demonstrated how the consecutive condensation of benzene rings together with two-dimensional molecular arrangement and the position of the hydroxyl group modifies this equilibrium. The obtained results revealed that the prototropic rearrangement in the electronic ground state is not thermodynamically less probable between two neighbouring condensed benzene rings. The keto form is favoured in linear polycyclic aromatic hydrocarbons for substituted central moieties. The angular molecular structure has the opposite effect. Based on the theoretical energies calculated for room temperature, the tautomerisation pKT constants and acidity pKa constants for enols as well as corresponding keto-tautomers were predicted and compared with available experimental values for the water environment. Finally, the possible experimental consequences in respect to the chemical reactivity of studied tautomers were discussed.
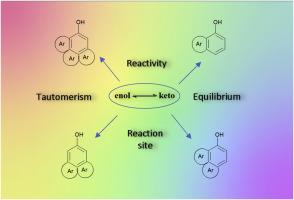
羟基取代茴香的酮烯醇同分异构:理论研究和实验结果
本文研究了苯酚、萘酚和选定的 29 种羟基取代的多环芳烃(分为 4 种结构类型)中出现的烯醇和酮同系物之间的化学平衡。使用密度泛函理论结合溶剂连续模型计算了反应吉布斯能。我们证明了苯环的连续缩合、二维分子排列和羟基的位置是如何改变这种平衡的。研究结果表明,在两个相邻的缩合苯环之间,电子基态的原向重排在热力学上的可能性并不低。在线性多环芳烃中,取代中心分子的酮形式更受青睐。角分子结构具有相反的效果。根据室温下计算出的理论能量,预测了烯醇以及相应酮-同分异构体的同分异构 pKT 常数和酸度 pKa 常数,并将其与水环境下的现有实验值进行了比较。最后,还讨论了所研究的同分异构体的化学反应性可能产生的实验结果。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


