{"title":"Superimposing Ligands with a Ligand Overlay as an Alternate Topology Model for λ-Dynamics-Based Calculations","authors":"Michael P. Liesen, and , Jonah Z. Vilseck*, ","doi":"10.1021/acs.jpcb.4c0480510.1021/acs.jpcb.4c04805","DOIUrl":null,"url":null,"abstract":"<p >Alchemical free energy (AFE) calculations can predict binding affinity changes as a function of structural modifications and have become powerful tools for lead optimization and drug discovery. Central to the setup and performance of AFE calculations is the manner of mapping alchemical transformations, known as the topology model. Single, dual, and hybrid topology models have been used with various AFE methods in the field. In recent works, λ-dynamics (λD) free energy calculations, specifically, have preferred the use of a hybrid multiple topology (HMT) for sampling multiple ligand perturbations. In this work, we evaluate a new topology method called ligand overlay (LO) for use with λD-based calculations, including the recently introduced λ-dynamics with a bias-updated Gibbs sampling (LaDyBUGS) approach. LO is a full multiple topology model that allows entire ligands to be sampled and restrained within a λ-dynamics framework. Relative binding free energies were computed with HMT or LO topology models with LaDyBUGS for 45 ligands across five protein benchmark systems. An overall Pearson <i>R</i> correlation of 0.98 and mean unsigned error of 0.32 kcal/mol were observed, suggesting that LO is a viable alternative topology model for λD-based calculations. We discuss the merits of using an HMT or LO model for future ligand studies with λD or LaDyBUGS calculations.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":"128 46","pages":"11359–11368 11359–11368"},"PeriodicalIF":2.9000,"publicationDate":"2024-11-08","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcb.4c04805","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
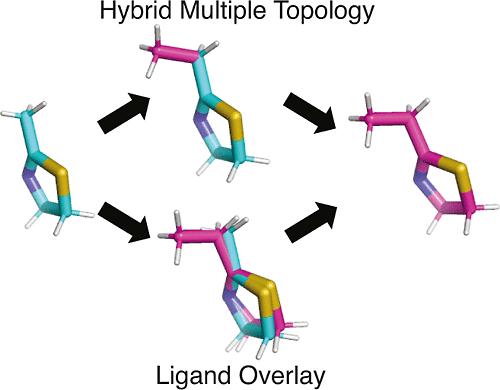
Alchemical free energy (AFE) calculations can predict binding affinity changes as a function of structural modifications and have become powerful tools for lead optimization and drug discovery. Central to the setup and performance of AFE calculations is the manner of mapping alchemical transformations, known as the topology model. Single, dual, and hybrid topology models have been used with various AFE methods in the field. In recent works, λ-dynamics (λD) free energy calculations, specifically, have preferred the use of a hybrid multiple topology (HMT) for sampling multiple ligand perturbations. In this work, we evaluate a new topology method called ligand overlay (LO) for use with λD-based calculations, including the recently introduced λ-dynamics with a bias-updated Gibbs sampling (LaDyBUGS) approach. LO is a full multiple topology model that allows entire ligands to be sampled and restrained within a λ-dynamics framework. Relative binding free energies were computed with HMT or LO topology models with LaDyBUGS for 45 ligands across five protein benchmark systems. An overall Pearson R correlation of 0.98 and mean unsigned error of 0.32 kcal/mol were observed, suggesting that LO is a viable alternative topology model for λD-based calculations. We discuss the merits of using an HMT or LO model for future ligand studies with λD or LaDyBUGS calculations.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


