Dispersion-corrected DFT calculations and dynamic molecular simulations to investigate conformational stability of Lidocaine towards β-CD and HP-β-CD
IF 2.7
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
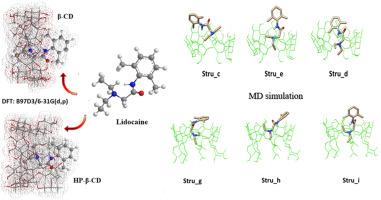
Lidocaine (LDC) is one of the most important local anaesthesia compounds (LAs), designated to treat acute and chronic pain, especially in clinical applications. In the purpose to improve its lower solubility and bioavailability, numerous researches have been conducted to study the exact mode of association between the LDC molecule and cyclodextrins as drug carriers. Although, the reported structural details on LDC/β-CD and LDC/HP-β-CD inclusion complexes remain largely unexplored. The LDC molecule presents different spatial arrangements inside the hydrophobic cavities of the above-mentioned hosts; either the phenyl moiety or the diethylamino part is totally inserted. Hence, in the present work, we attempt to deepen our understanding about conformational preferences on the binding modes of LDC by investigating the quantum mechanical approach results.
The PM3 method combined with the pure corrected functional B97D3 revealed the tendency of LDC to enter its diethylamino inside the host, leaving the rest of molecule externally, and consequently form an inclusion complex with HP-β-CD more stable than with the native β-CD by approximately 12 kcal mol−1.
The probability of partial insertion of LDC is further ascertained by MD simulations investigation running for 500 ns. The trajectory analysis of MD process showed that the diethyl amino fragment is accommodated inside the HP-β-CD's cavity for a significant period (82 % of the simulation time), while it is estimated to be 78 % in the case of LDC/β-CD complex.
Moreover, the wave function analysis, based on QTAIM, Reduced Density Gradient (RDG) and 2D Fingerprint, illustrated NCIs interactions and sustained the contribution of numerous van der Waals forces and weaker H-bonds interactions in the stability of studied ICs.
通过分散校正 DFT 计算和动态分子模拟研究利多卡因对 β-CD 和 HP-β-CD 的构象稳定性。
利多卡因(LDC)是最重要的局部麻醉化合物(LAs)之一,被指定用于治疗急性和慢性疼痛,尤其是在临床应用中。为了提高利多卡因的低溶解度和生物利用度,人们开展了大量研究,探讨利多卡因分子与作为药物载体的环糊精之间的确切结合模式。尽管如此,有关 LDC/β-CD 和 LDC/HP-β-CD 包合物结构细节的报道在很大程度上仍未得到证实。LDC 分子在上述载体的疏水空腔中呈现出不同的空间排列;苯基或二乙基氨基部分完全插入其中。因此,在本研究中,我们试图通过研究量子力学方法的结果,加深对 LDC 结合模式构象偏好的理解。PM3 方法结合纯校正函数 B97D3 发现,LDC 倾向于将其二乙胺进入宿主内部,而将分子的其余部分留在外部,因此与 HP-β-CD 形成的包合物比与原生 β-CD 形成的包合物稳定约 12 kcal mol-1。运行 500 ns 的 MD 模拟研究进一步确定了 LDC 部分插入的概率。MD 过程的轨迹分析表明,二乙基氨基片段在 HP-β-CD 的空腔中停留了相当长的时间(模拟时间的 82%),而在 LDC/β-CD 复合物的情况下,这一比例估计为 78%。此外,基于 QTAIM、还原密度梯度 (RDG) 和二维指纹的波函数分析表明了 NCIs 的相互作用,并证实了大量范德华力和较弱的 H 键相互作用对所研究 ICs 的稳定性的贡献。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


