Choice of Layering and Band Alignment in 2D Heterostructures
IF 3.3
3区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Heterostructures are ubiquitous in many optoelectronic devices and as photocatalysts. One of the key features of a heterojunction is proper band alignment between the two materials. Estimation of the correct relative band positions with density functional theory (DFT)-based electronic structure calculations is often constrained by the accuracy and cost associated with the various DFT functionals. In this study, we introduce a novel computational approach that achieves band alignments closely matching experimental results with the widely used PBE functional. We specifically examine the well-documented MoO3/MoS2 system, a type-II heterojunction. In our setup, the MoS2 layers are kept as they are, but for MoO3, the individual layers are chosen differently. These alternative layers have higher surface energy, and hence, the band edges are higher than those of the conventional layers. This shift in band edges of the alternative MoO3 layers changes the band alignment in the MoO3/MoS2 heterojunction from type-III to the experimentally observed type-II character. We also extend this computational strategy to additional systems, demonstrating its versatility and effectiveness.
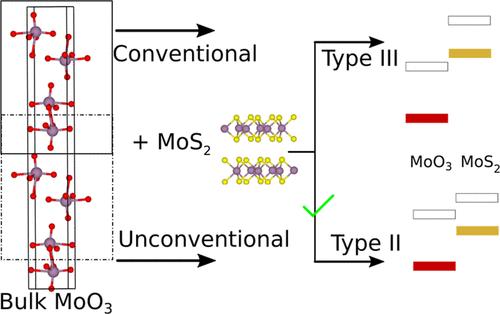
二维异质结构中层叠和带排列的选择
在许多光电设备和光催化剂中,异质结构无处不在。异质结的关键特征之一是两种材料之间的适当带排列。利用基于密度泛函理论(DFT)的电子结构计算来估计正确的相对能带位置,往往会受到各种 DFT 函数的精度和成本的限制。在本研究中,我们介绍了一种新颖的计算方法,它能利用广泛使用的 PBE 函数实现与实验结果接近的能带排列。我们特别研究了记录详实的 MoO3/MoS2 系统,这是一种 II 型异质结。在我们的设置中,MoS2 层保持原样,但对于 MoO3,单个层的选择有所不同。这些替代层具有更高的表面能,因此其带边高于传统层。替代 MoO3 层带边的这种偏移改变了 MoO3/MoS2 异质结的带排列,从 III 型变为实验观察到的 II 型。我们还将这一计算策略扩展到其他系统,证明了它的多功能性和有效性。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

The Journal of Physical Chemistry C
化学-材料科学:综合
CiteScore
6.50
自引率
8.10%
发文量
2047
审稿时长
1.8 months
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


