Unraveling the noncovalent interactions in a organic crystal using Quantum theory of atoms in molecules
IF 4
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
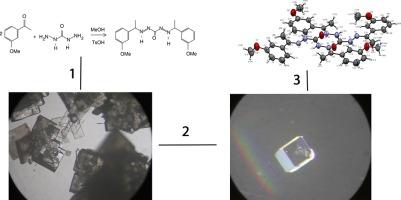
X-ray diffraction analysis, combined with the Quantum Theory of Atoms in Molecules (QTAIM), serves as a powerful tool for describing chemical bonding in real space for solids. By integrating theoretical and experimental data, a more accurate representation of atomic interactions including Van der Waals forces, hydrogen bonds, covalent, ionic, and metallic bonds is achieved. The analysis of noncovalent interactions through electronic density enables the identification of Lewis acid and base sites, while also revealing the directional ‘key-lock’ interactions that correspond to molecular recognition. The examination of critical points in the electron density and its derivatives allows for the characterization of the types of interactions present in crystal packing. This study focuses on the experimental and theoretical investigation of noncovalent interactions within a molecular crystal of a newly synthesized carbohydrazide derivative. The crystal structure was determined using X-ray single-crystal diffraction, and the crystallographic asymmetric unit was optimized via DFT, with the results compared to experimental data. Noncovalent interactions in real space such as Van der Waals forces, hydrogen bonds, and inter- and intramolecular steric repulsions were analyzed in terms of electron density and its derivatives. The QTAIM framework was applied to quantify the strength of these interactions, employing Voronoi deformation density and electron localization and delocalization indices for solids. The results presented in this work, using crystal engineering, reveal that derivatives of diurea compounds crystallize following a characteristic pattern that forms a synthon configuration. The strength of this interaction, quantified through QTAIM analysis of the electronic density, provides a deeper understanding of the chemistry of these compounds, both in terms of biological activity and coordination chemistry.
利用分子中原子的量子理论揭示有机晶体中的非共价相互作用
X 射线衍射分析与分子中原子的量子理论(QTAIM)相结合,是描述固体真实空间中化学键的有力工具。通过整合理论和实验数据,可以更准确地表示原子间的相互作用,包括范德华力、氢键、共价键、离子键和金属键。通过电子密度对非共价相互作用的分析,可以确定路易斯酸和碱的位点,同时还揭示了与分子识别相对应的定向 "键锁 "相互作用。通过对电子密度临界点及其衍生物的研究,可以确定晶体堆积中存在的相互作用类型。本研究的重点是对一种新合成的羧酰肼衍生物分子晶体中的非共价相互作用进行实验和理论研究。利用 X 射线单晶衍射确定了晶体结构,并通过 DFT 优化了晶体不对称单元,将结果与实验数据进行了比较。通过电子密度及其衍生物分析了实际空间中的非共价相互作用,如范德华力、氢键以及分子间和分子内的立体排斥。采用 QTAIM 框架,利用 Voronoi 变形密度和固体电子局域化和脱局域化指数来量化这些相互作用的强度。这项研究利用晶体工程学得出的结果表明,二脲化合物的衍生物结晶时遵循一种形成合子构型的特征模式。通过对电子密度进行 QTAIM 分析,量化了这种相互作用的强度,从而加深了对这些化合物在生物活性和配位化学方面的理解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊
Journal of Molecular Structure
化学-物理化学
CiteScore
7.10
自引率
15.80%
发文量
2384
审稿时长
45 days
期刊介绍:
The Journal of Molecular Structure is dedicated to the publication of full-length articles and review papers, providing important new structural information on all types of chemical species including:
• Stable and unstable molecules in all types of environments (vapour, molecular beam, liquid, solution, liquid crystal, solid state, matrix-isolated, surface-absorbed etc.)
• Chemical intermediates
• Molecules in excited states
• Biological molecules
• Polymers.
The methods used may include any combination of spectroscopic and non-spectroscopic techniques, for example:
• Infrared spectroscopy (mid, far, near)
• Raman spectroscopy and non-linear Raman methods (CARS, etc.)
• Electronic absorption spectroscopy
• Optical rotatory dispersion and circular dichroism
• Fluorescence and phosphorescence techniques
• Electron spectroscopies (PES, XPS), EXAFS, etc.
• Microwave spectroscopy
• Electron diffraction
• NMR and ESR spectroscopies
• Mössbauer spectroscopy
• X-ray crystallography
• Charge Density Analyses
• Computational Studies (supplementing experimental methods)
We encourage publications combining theoretical and experimental approaches. The structural insights gained by the studies should be correlated with the properties, activity and/ or reactivity of the molecule under investigation and the relevance of this molecule and its implications should be discussed.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


