Geometrical features and chemical adsorptions of (Ag3Sn)n clusters
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
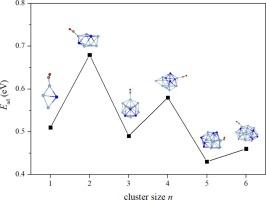
The Ag-Sn alloys are famous ancient intermetallics, with the Ag3Sn being a crucial component of the phase diagram. Recently, Ag3Sn nanoparticles showcase efficient catalytic CO oxidation capabilities. Here, structural features and stability of (Ag3Sn)n (n = 1–6) clusters are first analyzed in detail. The results reveal that structures of them evolve from cages to close-packed icosahedra, where Ag are distributed on cores and gradually aggregated, whereas Sn occupy edge positions and become dispersed. Moreover, the icosahedral (Ag3Sn)3 has a higher stability than that of its neighbors and can maintain the structural integrity at 700 K. The molecular orbitals reveal that the (Ag3Sn)3 has an electronic open-shell configuration of 1S21P61D102S21F1, which is confirmed by the density of states. Electrostatic potential surfaces show that (Ag3Sn)n have significant electron-deficient σ-hole regions at Ag sites, which can make C![]() O stretching frequencies and bond lengths have red-shifts. Adsorption energies between (Ag3Sn)n and CO display odd–even oscillations, ranging from (0.43–0.68) eV, and the direction of charge flows is from CO → clusters. Our work provides inferences to structure evolutions and adsorptions of the Ag3Sn alloy at the atomic level.
O stretching frequencies and bond lengths have red-shifts. Adsorption energies between (Ag3Sn)n and CO display odd–even oscillations, ranging from (0.43–0.68) eV, and the direction of charge flows is from CO → clusters. Our work provides inferences to structure evolutions and adsorptions of the Ag3Sn alloy at the atomic level.
(Ag3Sn)n 簇的几何特征和化学吸附作用
Ag-Sn合金是著名的古老金属间化合物,其中Ag3Sn是相图的重要组成部分。最近,Ag3Sn 纳米颗粒展示了高效的催化 CO 氧化能力。本文首次详细分析了 (Ag3Sn)n (n = 1-6)团簇的结构特征和稳定性。结果表明,它们的结构从笼状演化为紧密堆积的二十面体,其中银分布在核心并逐渐聚集,而锡占据边缘位置并变得分散。分子轨道显示,(Ag3Sn)3 的电子开壳构型为 1S21P61D102S21F1。静电位面显示(Ag3Sn)n在Ag位点上有明显的缺电子σ空穴区,这会使CO伸展频率和键长发生红移。(Ag3Sn)n与CO之间的吸附能呈现奇偶振荡,范围在(0.43-0.68)eV之间,电荷流的方向是从CO→簇。我们的工作提供了原子水平上 Ag3Sn 合金结构演变和吸附的推论。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


