Exploring the covalent inhibition mechanisms of inhibitors with two different warheads acting on SARS-CoV-2 Mpro by QM/MM simulations
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
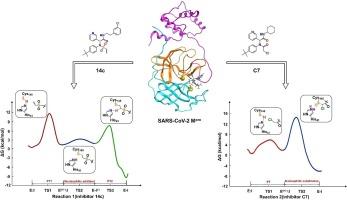
SARS-CoV-2 Mpro had been reported to react with covalent inhibitors 14c and C7, containing a vinyl sulfanilamide and chloromethylamide as the warheads, respectively. However, the detailed reaction mechanisms and warhead effects are still unclear. In this study, the initial state of the catalytic dyad Cys145/His41 is determined as neutral during the non-covalent binding, through MM-PBSA free energy calculations. The Potential of Mean Forces (PMFs) have been computed by QM/MM MD method, obtaining the free energy changes from reactants to products. Both reactions follow asynchronous mechanism. Except the similar proton transfer (PT1), subsequently, nucleophilic addition and an additional proton transfer (PT2) for 14c, while an SN2 nucleophilic substitution for C7 are underwent, respectively. The reaction activation barrier and free energy trends both are in consistent with the experimental IC50 results. Our research enhances understanding of the inhibitory mechanism, and provides insights for designing novel and more effective inhibitors targeting SARS-CoV-2 Mpro.
通过 QM/MM 模拟探索两种不同弹头的抑制剂对 SARS-CoV-2 Mpro 的共价抑制机制
据报道,SARS-CoV-2 Mpro 与共价抑制剂 14c 和 C7 发生反应,这两种抑制剂分别含有乙烯基磺酰胺和氯甲基酰胺作为弹头。然而,详细的反应机制和弹头效应仍不清楚。在本研究中,通过 MM-PBSA 自由能计算,确定了催化二元 Cys145/His41 在非共价结合过程中的初始状态为中性。通过 QM/MM MD 方法计算了平均力势(PMFs),得到了从反应物到产物的自由能变化。两个反应都遵循异步机理。除了类似的质子转移(PT1)外,14c 随后分别发生了亲核加成和额外的质子转移(PT2),而 C7 则发生了 SN2 亲核取代。反应活化势垒和自由能的变化趋势与 IC50 的实验结果一致。我们的研究加深了对抑制机理的理解,并为设计针对 SARS-CoV-2 Mpro 的更有效的新型抑制剂提供了启示。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


