First-principles study of aromatic amino acid encapsulation in single-walled BN and AlN nanotubes
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
Aromatic molecules exhibit strong non-covalent interactions with nanotubes, influencing their encapsulation properties. This study uses DFT calculations to explore the encapsulation of aromatic amino acids within zigzag (ZZ), chiral (R/S), and armchair (AC) single-walled aluminum nitride nanotubes (AlNNTs) and boron nitride nanotubes (BNNTs). The results reveal that zigzag AlNNTs exhibit the highest encapsulation affinity compared to other chiralities, while chiral BNNTs show enhanced encapsulation. Encapsulation energy decreases with increasing nanotube radius, indicating reduced affinity. Overall, the studied BNNTs demonstrate stronger encapsulation energy compared to AlNNTs. The bandgap energy of the encapsulated structures varies significantly with nanotube diameter and chirality. The physisorption process plays a major role in encapsulation, affecting the geometric and electronic properties of the nanotubes and enhancing the stability and efficacy of the encapsulated amino acids. These findings highlight the potential of these nanostructures for advanced applications, including targeted drug delivery and molecular sensing.
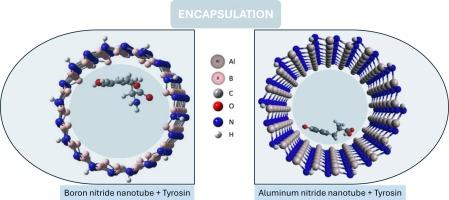
单壁 BN 和 AlN 纳米管中芳香族氨基酸封装的第一性原理研究
芳香族分子与纳米管之间表现出强烈的非共价相互作用,从而影响了纳米管的封装特性。本研究利用 DFT 计算探讨了芳香族氨基酸在人字形 (ZZ)、手性 (R/S) 和扶手椅 (AC) 单壁氮化铝纳米管 (AlNNT) 和氮化硼纳米管 (BNNT) 中的封装。结果表明,与其他手性相比,人字形氮化铝纳米管的封装亲和力最高,而手性氮化硼纳米管的封装能力更强。封装能随着纳米管半径的增加而降低,这表明亲和力降低了。总体而言,与 AlNNT 相比,所研究的 BNNT 具有更强的封装能。封装结构的带隙能随纳米管直径和手性的变化而显著不同。物理吸附过程在封装过程中发挥了重要作用,影响了纳米管的几何和电子特性,提高了封装氨基酸的稳定性和功效。这些发现凸显了这些纳米结构在靶向药物输送和分子传感等先进应用领域的潜力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


