Research on N, Ne, and P adsorption on boron-germanene nanoribbons for nano sensor applications
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
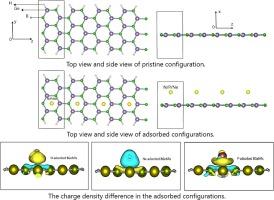
The study investigated the adsorption of N, P, and Ne atoms on boron-germanene nanoribbons (BGeNRs) using density functional theory (DFT) and the Vienna Ab initio Simulation Package (VASP). Results indicated that both the pristine and adsorbed configurations exhibited metallic behavior. While the pristine and Ne-adsorbed configurations were nonmagnetic, the N- and P-adsorbed configurations displayed magnetic moments of 1.81 μB and 1.21 μB, respectively. The N-adsorbed configuration had the lowest adsorption energy, whereas the Ne-adsorbed configuration exhibited a positive adsorption energy. Multi-orbital hybridization analysis revealed that hybridization processes predominantly occurred in the conduction band at energy levels corresponding to the σ bond. Charge density difference analysis showed significant charge transfer between the substrate and the adsorbed elements. Additionally, optical properties, including the real and imaginary parts of the dielectric function, absorption coefficient, and electron-hole density, were systematically examined to highlight the variations. The findings underscore the potential application of BGeNR materials in nanosensors.
用于纳米传感器应用的硼-锗纳米带对 N、Ne 和 P 的吸附研究
该研究利用密度泛函理论(DFT)和维也纳 Ab initio 仿真软件包(VASP)研究了 N、P 和 Ne 原子在硼-锗纳米带(BGeNRs)上的吸附。结果表明,原始构型和吸附构型都表现出金属特性。原始构型和Ne吸附构型没有磁性,而N和P吸附构型的磁矩分别为1.81 μB和1.21 μB。N 吸附构型的吸附能最低,而 Ne 吸附构型的吸附能为正值。多轨道杂化分析表明,杂化过程主要发生在导带中与σ键对应的能级上。电荷密度差分析表明,基底和吸附元素之间存在明显的电荷转移。此外,还系统地研究了光学特性,包括介电常数的实部和虚部、吸收系数和电子-空穴密度,以突出这些变化。这些发现强调了 BGeNR 材料在纳米传感器中的潜在应用。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


