The role of intra and intermolecular interactions on the conformational dynamics of 2-halo-1-phenylpropanols: Structure and solvent effects
IF 4
2区 化学
Q2 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
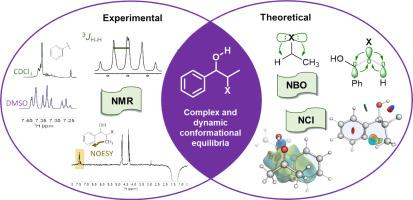
Experimental and theoretical 3JH![]() H scalar coupling constants allowed the identification of the conformational landscape of erythro (eX) and threo (tX) 2-halo-1-phenylpropanols (halo = F, Cl, and Br). NMR scalar coupling constants captured dynamics of the O
H scalar coupling constants allowed the identification of the conformational landscape of erythro (eX) and threo (tX) 2-halo-1-phenylpropanols (halo = F, Cl, and Br). NMR scalar coupling constants captured dynamics of the O![]() C-C-X dihedral and OH rotameric states, in a dynamic and solvent-dependent equilibrium. The erythro series revealed a particular halogen-dependent equilibrium, which showed different sensitivity to the media, especially in acetone, where the eF populations were completely shifted. At the same time, threo showed a highly solvent-sensitive equilibrium. NBO calculations showed the importance of electron delocalization over steric and electrostatic effects to stabilize the preferred synclinal conformer in both diastereomers. A Principal Component Analysis (PCA) on the NBO stabilization energies pointed to a complex mixture of electronic delocalization happening simultaneously. Hyperconjugative interactions are significant, but they are not the only important effect. Non-covalent interactions were also identified through NCI surfaces. Hydrogen bonds and intramolecular C-X···π and C
C-C-X dihedral and OH rotameric states, in a dynamic and solvent-dependent equilibrium. The erythro series revealed a particular halogen-dependent equilibrium, which showed different sensitivity to the media, especially in acetone, where the eF populations were completely shifted. At the same time, threo showed a highly solvent-sensitive equilibrium. NBO calculations showed the importance of electron delocalization over steric and electrostatic effects to stabilize the preferred synclinal conformer in both diastereomers. A Principal Component Analysis (PCA) on the NBO stabilization energies pointed to a complex mixture of electronic delocalization happening simultaneously. Hyperconjugative interactions are significant, but they are not the only important effect. Non-covalent interactions were also identified through NCI surfaces. Hydrogen bonds and intramolecular C-X···π and C![]() H···π interactions were proved to act differently in the two diastereomers, affecting their equilibria in different ways. Nuclear Overhauser (NOE) NMR experiments point to an intramolecular C
H···π interactions were proved to act differently in the two diastereomers, affecting their equilibria in different ways. Nuclear Overhauser (NOE) NMR experiments point to an intramolecular C![]() H···π contact, while 1H NMR of the aromatic hydrogens evidence an intermolecular effect of acetone and DMSO on the phenyl ring. DFT with explicit solvation shows a solvent shell favoring intermolecular C
H···π contact, while 1H NMR of the aromatic hydrogens evidence an intermolecular effect of acetone and DMSO on the phenyl ring. DFT with explicit solvation shows a solvent shell favoring intermolecular C![]() H···π contacts, in agreement with the experiments. This thorough analysis revealed that intra- and intermolecular factors contribute to the preference for the synclinal conformer in the studied compounds.
H···π contacts, in agreement with the experiments. This thorough analysis revealed that intra- and intermolecular factors contribute to the preference for the synclinal conformer in the studied compounds.
分子内和分子间相互作用对 2-卤代-1-苯基丙醇构象动力学的作用:结构和溶剂效应
通过实验和理论 3JHH 标量耦合常数,确定了赤式(eX)和苏式(tX)2-卤代-1-苯基丙醇(卤代 = F、Cl 和 Br)的构象格局。核磁共振标量耦合常数捕捉到了 OC-C-X 二面体和羟基旋转态的动态,处于一种动态的、依赖于溶剂的平衡状态。赤藓系列显示了一种特殊的卤素依赖性平衡,它对介质的敏感性不同,尤其是在丙酮中,eF 种群完全偏移。与此同时,苏氨酸显示出对溶剂高度敏感的平衡。NBO 计算表明,在稳定两种非对映异构体的优选同环构象方面,电子析出的重要性超过了立体效应和静电效应。对 NBO 稳定化能量进行的主成分分析(PCA)表明,同时发生的电子脱定位作用是一种复杂的混合作用。超共轭相互作用非常重要,但并不是唯一的重要影响。通过 NCI 表面还发现了非共价相互作用。氢键和分子内的 C-X-π 和 CH-π 相互作用在两种非对映异构体中的作用不同,以不同的方式影响着它们的平衡。核奥弗斯(NOE)核磁共振实验表明了分子内的 CH---π 接触,而芳香族氢的 1H 核磁共振则证明了丙酮和 DMSO 对苯环的分子间作用。明确溶解的 DFT 显示,溶剂壳有利于分子间 CH---π 接触,这与实验结果一致。这项全面的分析表明,分子内和分子间因素导致了所研究化合物的合成构象的偏好。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊
Journal of Molecular Structure
化学-物理化学
CiteScore
7.10
自引率
15.80%
发文量
2384
审稿时长
45 days
期刊介绍:
The Journal of Molecular Structure is dedicated to the publication of full-length articles and review papers, providing important new structural information on all types of chemical species including:
• Stable and unstable molecules in all types of environments (vapour, molecular beam, liquid, solution, liquid crystal, solid state, matrix-isolated, surface-absorbed etc.)
• Chemical intermediates
• Molecules in excited states
• Biological molecules
• Polymers.
The methods used may include any combination of spectroscopic and non-spectroscopic techniques, for example:
• Infrared spectroscopy (mid, far, near)
• Raman spectroscopy and non-linear Raman methods (CARS, etc.)
• Electronic absorption spectroscopy
• Optical rotatory dispersion and circular dichroism
• Fluorescence and phosphorescence techniques
• Electron spectroscopies (PES, XPS), EXAFS, etc.
• Microwave spectroscopy
• Electron diffraction
• NMR and ESR spectroscopies
• Mössbauer spectroscopy
• X-ray crystallography
• Charge Density Analyses
• Computational Studies (supplementing experimental methods)
We encourage publications combining theoretical and experimental approaches. The structural insights gained by the studies should be correlated with the properties, activity and/ or reactivity of the molecule under investigation and the relevance of this molecule and its implications should be discussed.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


