Data augmentation with generative models improves detection of Non-B DNA structures
IF 7
2区 医学
Q1 BIOLOGY
引用次数: 0
Abstract
Non-B DNA structures, or flipons, are important functional elements that regulate a large spectrum of cellular programs. Experimental technologies for flipon detection are limited to the subsets that are active at the time of an experiment and cannot capture whole-genome functional set. Thus, the task of generating reliable whole-genome annotations of non-B DNA structures is put on deep learning models, however their quality depends on the available experimental data for training. The data augmentation approach as the combination of synthetic and real data is widely used in various fields. Deep generative models demonstrated promising results in data augmentation improving classifiers’ performance. Here we aimed at testing performance of diffusion models in comparison to other generative models in generating synthetic non-B DNA structures for data augmentation approach. We tested denoising diffusion probabilistic and implicit models (DDPM and DDIM), Wasserstein generative adversarial network (WGAN), vector quantised variational autoencoder (VQ-VAE) and showed that data augmentation improves the quality of classifiers. Diffusion models overall show the best results, but when considering three criteria of generative trilemma - quality of generated samples, diversity and sampling speed, we conclude that trade-off is possible between generative diffusion model and other architectures such as WGAN and VQ-VAE.
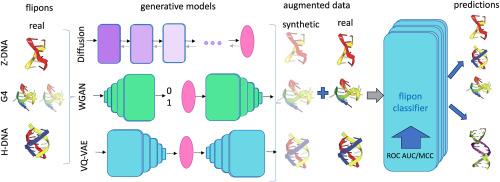
利用生成模型进行数据扩增可改进非 B 型 DNA 结构的检测。
非 B 型 DNA 结构或翻转子是调节大量细胞程序的重要功能元件。用于检测flipon的实验技术仅限于实验时活跃的子集,无法捕捉到全基因组功能集。因此,生成可靠的非 B DNA 结构全基因组注释的任务就落在了深度学习模型上,然而其质量取决于用于训练的可用实验数据。数据增强方法是合成数据和真实数据的结合,被广泛应用于各个领域。深度生成模型在数据扩增方面取得了可喜的成果,提高了分类器的性能。在此,我们旨在测试扩散模型与其他生成模型在生成合成非 B DNA 结构的数据增强方法中的性能对比。我们测试了去噪扩散概率模型和隐含模型(DDPM 和 DDIM)、瓦瑟斯坦生成对抗网络(WGAN)、向量量化变异自动编码器(VQ-VAE),结果表明数据扩增提高了分类器的质量。扩散模型总体上显示出最佳结果,但考虑到生成三难的三个标准--生成样本的质量、多样性和采样速度,我们得出结论,在生成扩散模型和其他架构(如 WGAN 和 VQ-VAE)之间可以进行权衡。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computers in biology and medicine
工程技术-工程:生物医学
CiteScore
11.70
自引率
10.40%
发文量
1086
审稿时长
74 days
期刊介绍:
Computers in Biology and Medicine is an international forum for sharing groundbreaking advancements in the use of computers in bioscience and medicine. This journal serves as a medium for communicating essential research, instruction, ideas, and information regarding the rapidly evolving field of computer applications in these domains. By encouraging the exchange of knowledge, we aim to facilitate progress and innovation in the utilization of computers in biology and medicine.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


