Probing Functional Allosteric States and Conformational Ensembles of the Allosteric Protein Kinase States and Mutants: Atomistic Modeling and Comparative Analysis of AlphaFold2, OmegaFold, and AlphaFlow Approaches and Adaptations
Nishank Raisinghani, Mohammed Alshahrani, Grace Gupta, Hao Tian, Sian Xiao, Peng Tao and Gennady Verkhivker*,
{"title":"Probing Functional Allosteric States and Conformational Ensembles of the Allosteric Protein Kinase States and Mutants: Atomistic Modeling and Comparative Analysis of AlphaFold2, OmegaFold, and AlphaFlow Approaches and Adaptations","authors":"Nishank Raisinghani, Mohammed Alshahrani, Grace Gupta, Hao Tian, Sian Xiao, Peng Tao and Gennady Verkhivker*, ","doi":"10.1021/acs.jpcb.4c0498510.1021/acs.jpcb.4c04985","DOIUrl":null,"url":null,"abstract":"<p >This study reports a comprehensive analysis and comparison of several AlphaFold2 adaptations and OmegaFold and AlphaFlow approaches in predicting distinct allosteric states, conformational ensembles, and mutation-induced structural effects for a panel of state-switching allosteric ABL mutants. The results revealed that the proposed AlphaFold2 adaptation with randomized alanine sequence scanning can generate functionally relevant allosteric states and conformational ensembles of the ABL kinase that qualitatively capture a unique pattern of population shifts between the active and inactive states in the allosteric ABL mutants. Consistent with the NMR experiments, the proposed AlphaFold2 adaptation predicted that G269E/M309L/T408Y mutant could induce population changes and sample a significant fraction of the fully inactive I<sub>2</sub> form which is a low-populated, high-energy state for the wild-type ABL protein. We also demonstrated that other ABL mutants G269E/M309L/T334I and M309L/L320I/T334I that introduce a single activating T334I mutation can reverse equilibrium and populate exclusively the active ABL form. While the precise quantitative predictions of the relative populations of the active and various hidden inactive states in the ABL mutants remain challenging, our results provide evidence that AlphaFold2 adaptation with randomized alanine sequence scanning can adequately detect a spectrum of the allosteric ABL states and capture the equilibrium redistributions between structurally distinct functional ABL conformations. We further validated the robustness of the proposed AlphaFold2 adaptation for predicting the unique inactive architecture of the BSK8 kinase and structural differences between ligand-unbound apo and ATP-bound forms of BSK8. The results of this comparative study suggested that AlpahFold2, OmegaFold, and AlphaFlow approaches may be driven by structural memorization of existing protein folds and are strongly biased toward predictions of the thermodynamically stable ground states of the protein kinases, highlighting limitations and challenges of AI-based methodologies in detecting alternative functional conformations, accurate characterization of physically significant conformational ensembles, and prediction of mutation-induced allosteric structural changes.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":"128 45","pages":"11088–11107 11088–11107"},"PeriodicalIF":2.8000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcb.4c04985","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
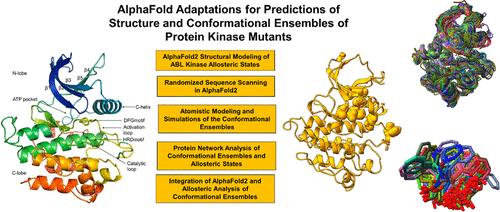
This study reports a comprehensive analysis and comparison of several AlphaFold2 adaptations and OmegaFold and AlphaFlow approaches in predicting distinct allosteric states, conformational ensembles, and mutation-induced structural effects for a panel of state-switching allosteric ABL mutants. The results revealed that the proposed AlphaFold2 adaptation with randomized alanine sequence scanning can generate functionally relevant allosteric states and conformational ensembles of the ABL kinase that qualitatively capture a unique pattern of population shifts between the active and inactive states in the allosteric ABL mutants. Consistent with the NMR experiments, the proposed AlphaFold2 adaptation predicted that G269E/M309L/T408Y mutant could induce population changes and sample a significant fraction of the fully inactive I2 form which is a low-populated, high-energy state for the wild-type ABL protein. We also demonstrated that other ABL mutants G269E/M309L/T334I and M309L/L320I/T334I that introduce a single activating T334I mutation can reverse equilibrium and populate exclusively the active ABL form. While the precise quantitative predictions of the relative populations of the active and various hidden inactive states in the ABL mutants remain challenging, our results provide evidence that AlphaFold2 adaptation with randomized alanine sequence scanning can adequately detect a spectrum of the allosteric ABL states and capture the equilibrium redistributions between structurally distinct functional ABL conformations. We further validated the robustness of the proposed AlphaFold2 adaptation for predicting the unique inactive architecture of the BSK8 kinase and structural differences between ligand-unbound apo and ATP-bound forms of BSK8. The results of this comparative study suggested that AlpahFold2, OmegaFold, and AlphaFlow approaches may be driven by structural memorization of existing protein folds and are strongly biased toward predictions of the thermodynamically stable ground states of the protein kinases, highlighting limitations and challenges of AI-based methodologies in detecting alternative functional conformations, accurate characterization of physically significant conformational ensembles, and prediction of mutation-induced allosteric structural changes.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


