{"title":"What Is the Protonation State of Proteins in Crystals? Insights from Constant pH Molecular Dynamics Simulations","authors":"Noora Aho*, Gerrit Groenhof and Pavel Buslaev*, ","doi":"10.1021/acs.jpcb.4c0594710.1021/acs.jpcb.4c05947","DOIUrl":null,"url":null,"abstract":"<p >X-ray crystallography is an important technique to determine the positions of atoms in a protein crystal. However, because the native environment in which proteins function, is not a crystal, but a solution, it is not a priori clear if the crystal structure represents the functional form of the protein. Because the protein structure and function often depend critically on the pH, the question arises whether proton affinities are affected by crystallization. X-ray diffraction usually does not reveal protons, which makes it difficult to experimentally measure p<i>K</i><sub>a</sub> shifts in crystals. Here, we investigate whether this challenge can be addressed by performing in silico titration with constant pH molecular dynamics (MD) simulations. We compare the computed p<i>K</i><sub>a</sub> values of proteins between solution and crystal environment and analyze these differences in the context of molecular interactions. For the proteins considered in this work, p<i>K</i><sub>a</sub> shifts were mostly found for residues at the crystal interfaces, where the environment is more apolar in the crystal than in water. Although convergence was an obstacle, our simulations suggest that in principle it is possible to apply constant pH MD to protein crystals routinely and assess the effect of crystallization on protein function more systematically than with standard MD simulations. We also highlight technical challenges that need to be addressed to make MD simulations of crystals more reliable.</p>","PeriodicalId":60,"journal":{"name":"The Journal of Physical Chemistry B","volume":"128 45","pages":"11124–11133 11124–11133"},"PeriodicalIF":2.8000,"publicationDate":"2024-10-31","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry B","FirstCategoryId":"1","ListUrlMain":"https://pubs.acs.org/doi/10.1021/acs.jpcb.4c05947","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
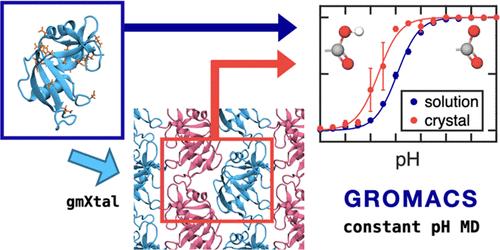
Abstract
X-ray crystallography is an important technique to determine the positions of atoms in a protein crystal. However, because the native environment in which proteins function, is not a crystal, but a solution, it is not a priori clear if the crystal structure represents the functional form of the protein. Because the protein structure and function often depend critically on the pH, the question arises whether proton affinities are affected by crystallization. X-ray diffraction usually does not reveal protons, which makes it difficult to experimentally measure pKa shifts in crystals. Here, we investigate whether this challenge can be addressed by performing in silico titration with constant pH molecular dynamics (MD) simulations. We compare the computed pKa values of proteins between solution and crystal environment and analyze these differences in the context of molecular interactions. For the proteins considered in this work, pKa shifts were mostly found for residues at the crystal interfaces, where the environment is more apolar in the crystal than in water. Although convergence was an obstacle, our simulations suggest that in principle it is possible to apply constant pH MD to protein crystals routinely and assess the effect of crystallization on protein function more systematically than with standard MD simulations. We also highlight technical challenges that need to be addressed to make MD simulations of crystals more reliable.
期刊介绍:
An essential criterion for acceptance of research articles in the journal is that they provide new physical insight. Please refer to the New Physical Insights virtual issue on what constitutes new physical insight. Manuscripts that are essentially reporting data or applications of data are, in general, not suitable for publication in JPC B.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


