In silico drug encapsulation using 2-hydroxypropyl-β-CD, tyrosine kinase and tyrosinase inhibition of dinuclear Cu(II) carboxylate complexes
IF 2.7
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
In recent years, copper carboxylate complexes have garnered significant interest for biological applications. This study focuses on 20 Cu(II) carboxylate complexes selected from our previous research. Due to the hydrophobic nature of these complexes, the 2-hydroxypropyl-β-cyclodextrin (2HPβCD) was employed as a carrier to reduce toxicity and increase solubility for controlling drug delivery. Monte Carlo calculations were performed to confirm the interaction between the optimized structures of Cu(II) complexes and 2HPβCD, forming a host-guest system. All the structures were simulated and optimized using DFT-D calculations in Material Studio 2017. The results indicated that a neutral medium is more favorable for the adsorption of these complexes into 2HPβCD. More negative binding energy values suggested strong and energetically favorable adsorption on 2HPβCD. Complexes 4, 5, and 7 exhibited the highest interaction, making them excellent candidates for drug delivery systems. DFT-D calculations were also used to investigate the release of complexes, revealing that complexes 5, 14, and 19 were difficult to release due to their lowest energy. In contrast, complexes 8, 9, and 16 were found to be most efficient to release due to weak non-covalent interactions with 2HPβCD as we can predict from binding energy obtained by DFT-D. No specific trend was observed in the interaction of the complexes with 2HPβCD. Additionally, the effects of these complexes on c-kit tyrosine kinase and Mushroom tyrosinase were studied by molecular docking. The results demonstrated that all the complexes interacted with the active site of respective receptors through hydrophobic interactions. Complexes containing 1,10-phenanthroline and 2,2-bipyrdine were identified as having a strong, spontaneous binding ability with receptors.
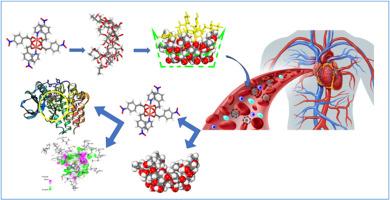
利用 2-hydroxypropyl-β-CD 进行药物包囊、双核羧酸铜(II)配合物的酪氨酸激酶和酪氨酸酶抑制作用的硅学研究。
近年来,羧酸铜配合物在生物应用方面引起了极大的兴趣。本研究侧重于从我们以前的研究中选出的 20 个羧酸铜(II)配合物。由于这些配合物具有疏水性,因此采用了 2-hydroxypropyl-β-cyclodextrin (2HPβCD) 作为载体,以降低毒性并增加溶解度,从而控制药物的输送。通过蒙特卡洛计算确认了 Cu(II)配合物与 2HPβCD 的优化结构之间的相互作用,形成了一个主-客系统。所有结构均使用 Material Studio 2017 中的 DFT-D 计算进行模拟和优化。结果表明,中性介质更有利于这些复合物吸附到 2HPβCD 中。结合能的负值越大,表明在 2HPβCD 上的吸附力越强,能量越有利。配合物 4、5 和 7 表现出最高的相互作用,使它们成为药物输送系统的理想候选物质。DFT-D 计算还用于研究配合物的释放,结果表明,配合物 5、14 和 19 由于能量最低而难以释放。相反,根据 DFT-D 计算得出的结合能,我们发现 8、9 和 16 复合物与 2HPβCD 的非共价相互作用较弱,因此释放效率最高。在复合物与 2HPβCD 的相互作用中没有观察到特定的趋势。此外,我们还通过分子对接研究了这些复合物对 c-kit 酪氨酸激酶和蘑菇酪氨酸酶的影响。结果表明,所有复合物都通过疏水作用与各自受体的活性位点相互作用。经鉴定,含有 1,10-菲罗啉和 2,2-联吡啶的复合物与受体具有很强的自发结合能力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


