Sabrina Setembre Batah, Andrea Jazel Rodriguez-Herrera, Maria Júlia Faci do Marco, Juliana Rocha Souza Chiappetto, Mariana Gatto, Simone Alves do Vale, Robson Aparecido Prudente, Amanda Piveta Schnepper, Robson Francisco Carvalho, João Paulo Facio Almeida, Tales Rubens de Nadai, Marcel Konigkam Santos, Li Siyuan Wada, José Baddini-Martinez, Danilo Tadao Wada, Andrea Antunes Cetlin, Vera Luiza Capelozzi, Bruno Guedes Baldi, Suzana Tanni, Rosane Duarte Achcar, Alexandre Todorovic Fabro
{"title":"Transcriptomic profiling reveals the dynamics of fibrotic progression-related gene expression into post-coronavirus disease 2019 pulmonary fibrosis","authors":"Sabrina Setembre Batah, Andrea Jazel Rodriguez-Herrera, Maria Júlia Faci do Marco, Juliana Rocha Souza Chiappetto, Mariana Gatto, Simone Alves do Vale, Robson Aparecido Prudente, Amanda Piveta Schnepper, Robson Francisco Carvalho, João Paulo Facio Almeida, Tales Rubens de Nadai, Marcel Konigkam Santos, Li Siyuan Wada, José Baddini-Martinez, Danilo Tadao Wada, Andrea Antunes Cetlin, Vera Luiza Capelozzi, Bruno Guedes Baldi, Suzana Tanni, Rosane Duarte Achcar, Alexandre Todorovic Fabro","doi":"10.1002/ctm2.70088","DOIUrl":null,"url":null,"abstract":"<p>Dear Editor,</p><p>Our study has identified a gene expression profile associated with the progression of coronavirus disease 2019 (COVID-19) to pulmonary fibrosis in a pro-fibrotic environment similar to that found in fibrosing interstitial lung diseases (f-ILDs). Briefly, we noted the common expression of 86 genes in post-COVID-19 pulmonary fibrosis (post-CPF) and f-ILDs, indicating their likely involvement in perpetuating pulmonary fibrosis through shared fibrotic pathways—confirmed by the in-situ expression of MUC5ac and WNT10a. Furthermore, an additional set of 31 genes exhibited common expression patterns between subacute COVID-19, the so-called organizing diffuse alveolar damage (ODAD), and CPF, as well as f-ILDs. Among those genes, MUC4 and KRT5 were confirmed by immunohistochemistry, suggesting their role as potential predictors for the early outcome of possible pulmonary fibrosis.</p><p>Post-CPF is a long-term complication diagnosed by clinical setting, pulmonary function tests and/or image examinations.<span><sup>1</sup></span> Initially, some COVID-19-infected patients develop acute respiratory distress syndrome (ARDS) during the exudative phase of DAD, marked by cytokine storm and immune cell recruitment.<span><sup>2</sup></span> Following the inflammatory peak and pneumocyte injury, myofibroblast activation triggers extracellular matrix (ECM) deposition, leading to ODAD-phase which typically restores to typical lung architecture. However, some patients progress to pulmonary fibrosis<span><sup>3</sup></span> with morphological changes that are driven by a complex pathophysiological sequence and dynamic gene expression shifts. In the end, the fibrotic outcome can resemble other f-ILDs. Identifying gene expression levels linked to the progression from ODAD to CPF is crucial for finding biomarkers for early diagnosis. Our study aimed to identify potential biomarkers in gene expression associated with fibrotic progression to CPF by analyzing the transcriptome of patients with ODAD, CPF, f-ILDs and controls.</p><p>As previously described by Batah et al.,<span><sup>4</sup></span> autopsies from the ODAD group revealed ODAD-phase with bronchiolar metaplasia, myxoid fibrosis, myofibroblastic activation and extensive alveolar septal thickening with collagen types I and III deposition (Table S1; Figure 1A–C). Meanwhile, after an average of 324.6 days following the initial positive nasopharyngeal swab for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), patients from the CPF group developed pulmonary fibrosis with bronchiolar metaplasia and parenchymal remodelling with increased collagen deposition, especially type I (suppinfo1; Figure 1D–F). Although the common remodelling profile, the differential gene expression (DGE) analysis between ODAD and CPF revealed distinct gene signatures (Figure S1A,B), Some of the top 20 DGEs reflect the manifestation of ARDS in ODAD patients (Figure S1C,D and Table S2). For example, the upregulation of SERPINE1 -fibrinolysis inhibitor- in ODAD suggests the accumulation of deposited fibrin as a better pathophysiological response to SARS-CoV-2.<span><sup>5</sup></span> However, as mentioned earlier, both groups revealed parenchymal remodelling, and the WP Lung Fibrosis gene set (GSEA systematic name M39477) highlighted only a few DGE between the groups: upregulated MMP9 and TERT and downregulated MUC5B and FGF1 in ODAD compared to CPF (Figure S1E and Table S3). Therefore, some fibrotic pathways might be present in both groups, suggesting a possible similarity with the transcriptomic profile of f-ILD patients.</p><p>In fact, our data revealed a significant similarity in gene expression among the groups compared to f-ILD, presenting dense fibrosis, architectural distortion, bronchiolar metaplasia, fibroblast foci and collagen types I and III deposition (Table S1 and Figure 1G–I). The similarity is seen by the overlap of cases in the paired DGE analyses between CPF versus f-ILD (Figure S2A,B) and ODAD versus f-ILD (Figure S3A,B). In both principal component analyses, f-ILD patients were divided into two distinct clusters: In Figure S2A, Cluster A is distinct from the other cases, while Cluster B is found among the CPF patients. Cluster A displays a bronchiolocentric remodelling pattern, whereas Cluster B exhibits a non-specific lesion pattern characterized by uniform thickening of the pulmonary interstitium, similar to that observed in CPF patients. Similarly, in Figure S3A, Cluster D has the same patients as Cluster A, who also exhibit bronchiolocentric ECM deposition; while Cluster E has a lesion pattern more similar to ODAD, which is also non-specific. This may suggest that the transcriptomic profiles of each cluster differently influence the pattern and intensity of ECM deposition, resulting in distinct forms of fibrosis depending on the genes expressed.</p><p>In addition, the top 20 DGE highlighted upregulation of COL1A1 and CDH23 in CPF versus f-ILD (Figure S2C,D and Table S4); and downregulation of miR-663a in ODAD versus f-ILD, which directly targets transforming growth factor (TGF)-β1<span><sup>6</sup></span> (Figure S3C-D and Table S5). However, the WP Lung Fibrosis gene set (GSEA systematic name M39477) did not reveal DGE between the groups, reinforcing the similarity in the fibrotic pathway pattern (Figures S2E and S3E; Tables S6 and S7).</p><p>To ascertain the similarity in the gene expression levels of CPF and ODAD with f-ILD, we performed the DE analysis of each group against CTR-minimally altered pulmonary parenchyma with significantly lower collagen fibre deposition compared to the other groups (Table S1 and Figure 1J–M). A Venn diagram was generated from the DGE results (ODAD vs. CTR, CPF vs. CTR and f-ILD vs. CTR) to highlight the overlapping up and downregulated genes. As a result, 86 genes (71 upregulated, 15 downregulated) were commonly expressed in CPF and f-ILD (Table S8 and Figure 2). The distinctiveness of these genes lies in their absence from the ODAD group, suggesting transcription under chronic injury conditions without an active injurious stimulus, which might be considered as “fibrosis-related genes”. These genes likely play a pivotal role in sustaining fibrosis in a chronic and advanced state.</p><p>Indeed, the upregulated Fibrosis-related genes show significant enrichment in pathways related to WNT signalling (Figure 3A,B), suggesting a potential role in the progression of post-CPF, possibly by promoting fibrosis and epithelial-mesenchymal transition (EMT).<span><sup>7</sup></span> Additionally, the Cadherin signalling pathway (Figure 3B) is also significantly enriched, further supporting its involvement in EMTs. To confirm the gene expression findings (Figure S4A–D), we selected two genes from the group of 86, MUC5ac and WNT10a, both of which are already widely used in diagnostic immunohistochemistry (IHC). We observed higher expression of goblet cell hyperplasia MUC5ac+ in CPF and f-ILD compared to the other groups (Figure 3C,D, <i>p</i> < .05), as reported in other studies<span><sup>8</sup></span>; and higher in-situ presence of WNT10a+ in squamous cell metaplasia in CPF and f-ILD (Figure 3E,F, <i>p</i> < .05), consistent with its role in pulmonary fibrosis as described in other studies.<span><sup>7</sup></span></p><p>The Venn diagram also emphasized 31 genes (23 upregulated and eight downregulated) commonly expressed across the three groups (ODAD, CPF and f-ILD) (Table S9 and Figure 2). These genes likely contribute to the transition from the early fibrotic stage in ODAD to the more chronic fibrotic injury observed in CPF. Their continued presence in well-established fibrosis (f-ILD) underscores their importance in fibrosis progression, leading us to classify them as “progression-related genes.” The enrichment analysis of the upregulated genes from this list revealed a significant association with the transcription factors SMAD2 and SMAD3 (Figure 4A), which are central to TGF-β signalling, a key pathway in EMT and the advancement of fibrotic diseases, including post-CPF. Additionally, these genes showed a strong link to the “innate immunity evasion and cell-specific immune response” pathway associated with SARS-CoV-2 infections (Figure 4B). This pathway is crucial for understanding how the virus evades the host immune responses, potentially influencing the development of severe conditions like post-CPF by disrupting immune detection and altering cell cycle regulation.</p><p>To further validate the gene expression findings (Figure S4E–J), we selected two additional markers, KRT5 and MUC4, both widely used in diagnostic IHC. Increased in-situ expression of the epithelial marker KRT5+ in basal cell hyperplasia was observed in all groups compared to CTR (Figure 4C,D, <i>p</i> < .05) consistent with squamous metaplasia reported in COVID-19 cases.<span><sup>9</sup></span> Similarly, elevated in-situ MUC4+ expression in bronchial goblet cell hyperplasia (Figure 4E,F) was observed, consistent with findings in idiopathic pulmonary fibrosis (IPF).<span><sup>10</sup></span> The authors addressed MUC4 as part of the TGF-β1 canonical pathway, suggesting its role in the fibrotic microenvironment.</p><p>In conclusion, our study identifies a gene expression profile associated with COVID-19 progression to pulmonary fibrosis, revealing shared pathways with f-ILDs. In this way, the KRT5+ cells impair epithelial repair, while MUC4 overexpression exacerbates inflammation and tissue damage, restricting proper regeneration and promoting pulmonary fibrosis. MUC5AC maintains inflammation through mucus hypersecretion, and WNT10a drives fibroblast proliferation and ECM production. Continuous activation of the Wnt pathway by WNT10a contributes to tissue stiffness, which is crucial for sustaining pulmonary fibrosis. Furthermore, we demonstrate increased transcriptomic and in-situ expression of MUC4 and KRT5, suggesting their pivotal role in fibrosis progression and potential as early diagnostic biomarkers in transbronchial biopsy samples. However, a limitation of our study is that it captures gene expression at a single time-point, and longitudinal studies are needed to track gene expression changes from the acute infection phase to fibrosis development. Additionally, further validation of these biomarkers in larger cohorts, as well as investigation into their underlying mechanisms, is necessary given their significant potential for clinical practice as immunohistochemical markers for early intervention in fibrotic diseases.</p><p>Conceptualization: Sabrina Setembre Batah and Alexandre Todorovic Fabro; Formal analysis: Sabrina Setembre Batah, Amanda Piveta Schnepper, Robson Francisco Carvalho and João Paulo Facio Almeida; Funding acquisition: Sabrina Setembre Batah and Alexandre Todorovic Fabro; Investigation: Sabrina Setembre Batah, Juliana Rocha Souza Chiappetto, Tales Rubens de Nadai, Marcel Konigkam Santos, Li Siyuan Wada, Danilo Tadao Wada, Andrea Antunes Cetlin, Bruno Guedes Baldi and Suzana Tanni; Methodology: Sabrina Setembre Batah, Andrea Jazel Rodriguez-Herrera, Maria Júlia Faci do Marco, Juliana Rocha Souza Chiappetto, Mariana Gatto, Simone Alves do Vale, Robson Aparecido Prudente, Amanda Piveta Schnepper and João Paulo Facio Almeida; Project administration: Alexandre Todorovic Fabro; Resources: Robson Francisco Carvalho, José Baddini-Martinez, Bruno Guedes Baldi, Suzana Tanni and Alexandre Todorovic Fabro; Software: Sabrina Setembre Batah, Amanda Piveta Schnepper, Robson Francisco Carvalho and João Paulo Facio Almeida; Supervision: Vera Luiza Capelozzi, Rosane Duarte Achcar and Alexandre Todorovic Fabro; Validation: Sabrina Setembre Batah, Robson Francisco Carvalho and Alexandre Todorovic Fabro; Visualization: José Baddini-Martinez, Vera Luiza Capelozzi, Rosane Duarte Achcar and Alexandre Todorovic Fabro; Writing—original draft: Sabrina Setembre Batah and Alexandre Todorovic Fabro; Writing—review & editing: Sabrina Setembre Batah, Robson Francisco Carvalho, José Baddini-Martinez, Vera Luiza Capelozzi, Suzana Tanni, Rosane Duarte Achcar and Alexandre Todorovic Fabro.</p><p>The authors declare no conflict of interest.</p><p>This research was supported by São Paulo Research Foundation (Fapesp 22/02821-0; 20/13370-4; 23/10186-6; 23/04199-8; 21/09024-6; 23/10186-6; 23/04199-8; 23/10184-3) and by the National Council for Scientific and Technological Development (CNPq 310415/2021-7).</p><p>The Research Ethics Committee approved this study, and written informed consent was waived (CAAE: 43040920.0.0000.5440, 03737018.6.0000.5440 and 65315822.0.0000.5411).</p>","PeriodicalId":10189,"journal":{"name":"Clinical and Translational Medicine","volume":"14 11","pages":""},"PeriodicalIF":7.9000,"publicationDate":"2024-11-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11560857/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Clinical and Translational Medicine","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/ctm2.70088","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MEDICINE, RESEARCH & EXPERIMENTAL","Score":null,"Total":0}
引用次数: 0
Abstract
Dear Editor,
Our study has identified a gene expression profile associated with the progression of coronavirus disease 2019 (COVID-19) to pulmonary fibrosis in a pro-fibrotic environment similar to that found in fibrosing interstitial lung diseases (f-ILDs). Briefly, we noted the common expression of 86 genes in post-COVID-19 pulmonary fibrosis (post-CPF) and f-ILDs, indicating their likely involvement in perpetuating pulmonary fibrosis through shared fibrotic pathways—confirmed by the in-situ expression of MUC5ac and WNT10a. Furthermore, an additional set of 31 genes exhibited common expression patterns between subacute COVID-19, the so-called organizing diffuse alveolar damage (ODAD), and CPF, as well as f-ILDs. Among those genes, MUC4 and KRT5 were confirmed by immunohistochemistry, suggesting their role as potential predictors for the early outcome of possible pulmonary fibrosis.
Post-CPF is a long-term complication diagnosed by clinical setting, pulmonary function tests and/or image examinations.1 Initially, some COVID-19-infected patients develop acute respiratory distress syndrome (ARDS) during the exudative phase of DAD, marked by cytokine storm and immune cell recruitment.2 Following the inflammatory peak and pneumocyte injury, myofibroblast activation triggers extracellular matrix (ECM) deposition, leading to ODAD-phase which typically restores to typical lung architecture. However, some patients progress to pulmonary fibrosis3 with morphological changes that are driven by a complex pathophysiological sequence and dynamic gene expression shifts. In the end, the fibrotic outcome can resemble other f-ILDs. Identifying gene expression levels linked to the progression from ODAD to CPF is crucial for finding biomarkers for early diagnosis. Our study aimed to identify potential biomarkers in gene expression associated with fibrotic progression to CPF by analyzing the transcriptome of patients with ODAD, CPF, f-ILDs and controls.
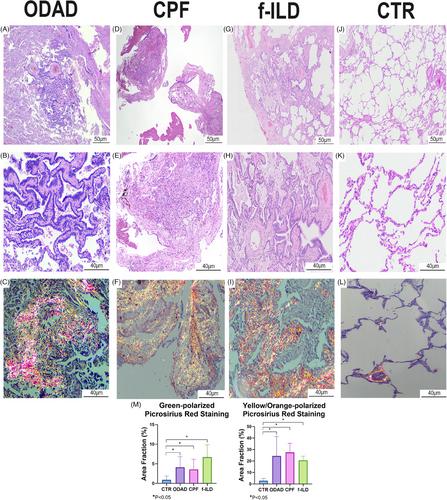
As previously described by Batah et al.,4 autopsies from the ODAD group revealed ODAD-phase with bronchiolar metaplasia, myxoid fibrosis, myofibroblastic activation and extensive alveolar septal thickening with collagen types I and III deposition (Table S1; Figure 1A–C). Meanwhile, after an average of 324.6 days following the initial positive nasopharyngeal swab for severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), patients from the CPF group developed pulmonary fibrosis with bronchiolar metaplasia and parenchymal remodelling with increased collagen deposition, especially type I (suppinfo1; Figure 1D–F). Although the common remodelling profile, the differential gene expression (DGE) analysis between ODAD and CPF revealed distinct gene signatures (Figure S1A,B), Some of the top 20 DGEs reflect the manifestation of ARDS in ODAD patients (Figure S1C,D and Table S2). For example, the upregulation of SERPINE1 -fibrinolysis inhibitor- in ODAD suggests the accumulation of deposited fibrin as a better pathophysiological response to SARS-CoV-2.5 However, as mentioned earlier, both groups revealed parenchymal remodelling, and the WP Lung Fibrosis gene set (GSEA systematic name M39477) highlighted only a few DGE between the groups: upregulated MMP9 and TERT and downregulated MUC5B and FGF1 in ODAD compared to CPF (Figure S1E and Table S3). Therefore, some fibrotic pathways might be present in both groups, suggesting a possible similarity with the transcriptomic profile of f-ILD patients.
In fact, our data revealed a significant similarity in gene expression among the groups compared to f-ILD, presenting dense fibrosis, architectural distortion, bronchiolar metaplasia, fibroblast foci and collagen types I and III deposition (Table S1 and Figure 1G–I). The similarity is seen by the overlap of cases in the paired DGE analyses between CPF versus f-ILD (Figure S2A,B) and ODAD versus f-ILD (Figure S3A,B). In both principal component analyses, f-ILD patients were divided into two distinct clusters: In Figure S2A, Cluster A is distinct from the other cases, while Cluster B is found among the CPF patients. Cluster A displays a bronchiolocentric remodelling pattern, whereas Cluster B exhibits a non-specific lesion pattern characterized by uniform thickening of the pulmonary interstitium, similar to that observed in CPF patients. Similarly, in Figure S3A, Cluster D has the same patients as Cluster A, who also exhibit bronchiolocentric ECM deposition; while Cluster E has a lesion pattern more similar to ODAD, which is also non-specific. This may suggest that the transcriptomic profiles of each cluster differently influence the pattern and intensity of ECM deposition, resulting in distinct forms of fibrosis depending on the genes expressed.
In addition, the top 20 DGE highlighted upregulation of COL1A1 and CDH23 in CPF versus f-ILD (Figure S2C,D and Table S4); and downregulation of miR-663a in ODAD versus f-ILD, which directly targets transforming growth factor (TGF)-β16 (Figure S3C-D and Table S5). However, the WP Lung Fibrosis gene set (GSEA systematic name M39477) did not reveal DGE between the groups, reinforcing the similarity in the fibrotic pathway pattern (Figures S2E and S3E; Tables S6 and S7).
To ascertain the similarity in the gene expression levels of CPF and ODAD with f-ILD, we performed the DE analysis of each group against CTR-minimally altered pulmonary parenchyma with significantly lower collagen fibre deposition compared to the other groups (Table S1 and Figure 1J–M). A Venn diagram was generated from the DGE results (ODAD vs. CTR, CPF vs. CTR and f-ILD vs. CTR) to highlight the overlapping up and downregulated genes. As a result, 86 genes (71 upregulated, 15 downregulated) were commonly expressed in CPF and f-ILD (Table S8 and Figure 2). The distinctiveness of these genes lies in their absence from the ODAD group, suggesting transcription under chronic injury conditions without an active injurious stimulus, which might be considered as “fibrosis-related genes”. These genes likely play a pivotal role in sustaining fibrosis in a chronic and advanced state.
Indeed, the upregulated Fibrosis-related genes show significant enrichment in pathways related to WNT signalling (Figure 3A,B), suggesting a potential role in the progression of post-CPF, possibly by promoting fibrosis and epithelial-mesenchymal transition (EMT).7 Additionally, the Cadherin signalling pathway (Figure 3B) is also significantly enriched, further supporting its involvement in EMTs. To confirm the gene expression findings (Figure S4A–D), we selected two genes from the group of 86, MUC5ac and WNT10a, both of which are already widely used in diagnostic immunohistochemistry (IHC). We observed higher expression of goblet cell hyperplasia MUC5ac+ in CPF and f-ILD compared to the other groups (Figure 3C,D, p < .05), as reported in other studies8; and higher in-situ presence of WNT10a+ in squamous cell metaplasia in CPF and f-ILD (Figure 3E,F, p < .05), consistent with its role in pulmonary fibrosis as described in other studies.7
The Venn diagram also emphasized 31 genes (23 upregulated and eight downregulated) commonly expressed across the three groups (ODAD, CPF and f-ILD) (Table S9 and Figure 2). These genes likely contribute to the transition from the early fibrotic stage in ODAD to the more chronic fibrotic injury observed in CPF. Their continued presence in well-established fibrosis (f-ILD) underscores their importance in fibrosis progression, leading us to classify them as “progression-related genes.” The enrichment analysis of the upregulated genes from this list revealed a significant association with the transcription factors SMAD2 and SMAD3 (Figure 4A), which are central to TGF-β signalling, a key pathway in EMT and the advancement of fibrotic diseases, including post-CPF. Additionally, these genes showed a strong link to the “innate immunity evasion and cell-specific immune response” pathway associated with SARS-CoV-2 infections (Figure 4B). This pathway is crucial for understanding how the virus evades the host immune responses, potentially influencing the development of severe conditions like post-CPF by disrupting immune detection and altering cell cycle regulation.
To further validate the gene expression findings (Figure S4E–J), we selected two additional markers, KRT5 and MUC4, both widely used in diagnostic IHC. Increased in-situ expression of the epithelial marker KRT5+ in basal cell hyperplasia was observed in all groups compared to CTR (Figure 4C,D, p < .05) consistent with squamous metaplasia reported in COVID-19 cases.9 Similarly, elevated in-situ MUC4+ expression in bronchial goblet cell hyperplasia (Figure 4E,F) was observed, consistent with findings in idiopathic pulmonary fibrosis (IPF).10 The authors addressed MUC4 as part of the TGF-β1 canonical pathway, suggesting its role in the fibrotic microenvironment.
In conclusion, our study identifies a gene expression profile associated with COVID-19 progression to pulmonary fibrosis, revealing shared pathways with f-ILDs. In this way, the KRT5+ cells impair epithelial repair, while MUC4 overexpression exacerbates inflammation and tissue damage, restricting proper regeneration and promoting pulmonary fibrosis. MUC5AC maintains inflammation through mucus hypersecretion, and WNT10a drives fibroblast proliferation and ECM production. Continuous activation of the Wnt pathway by WNT10a contributes to tissue stiffness, which is crucial for sustaining pulmonary fibrosis. Furthermore, we demonstrate increased transcriptomic and in-situ expression of MUC4 and KRT5, suggesting their pivotal role in fibrosis progression and potential as early diagnostic biomarkers in transbronchial biopsy samples. However, a limitation of our study is that it captures gene expression at a single time-point, and longitudinal studies are needed to track gene expression changes from the acute infection phase to fibrosis development. Additionally, further validation of these biomarkers in larger cohorts, as well as investigation into their underlying mechanisms, is necessary given their significant potential for clinical practice as immunohistochemical markers for early intervention in fibrotic diseases.
Conceptualization: Sabrina Setembre Batah and Alexandre Todorovic Fabro; Formal analysis: Sabrina Setembre Batah, Amanda Piveta Schnepper, Robson Francisco Carvalho and João Paulo Facio Almeida; Funding acquisition: Sabrina Setembre Batah and Alexandre Todorovic Fabro; Investigation: Sabrina Setembre Batah, Juliana Rocha Souza Chiappetto, Tales Rubens de Nadai, Marcel Konigkam Santos, Li Siyuan Wada, Danilo Tadao Wada, Andrea Antunes Cetlin, Bruno Guedes Baldi and Suzana Tanni; Methodology: Sabrina Setembre Batah, Andrea Jazel Rodriguez-Herrera, Maria Júlia Faci do Marco, Juliana Rocha Souza Chiappetto, Mariana Gatto, Simone Alves do Vale, Robson Aparecido Prudente, Amanda Piveta Schnepper and João Paulo Facio Almeida; Project administration: Alexandre Todorovic Fabro; Resources: Robson Francisco Carvalho, José Baddini-Martinez, Bruno Guedes Baldi, Suzana Tanni and Alexandre Todorovic Fabro; Software: Sabrina Setembre Batah, Amanda Piveta Schnepper, Robson Francisco Carvalho and João Paulo Facio Almeida; Supervision: Vera Luiza Capelozzi, Rosane Duarte Achcar and Alexandre Todorovic Fabro; Validation: Sabrina Setembre Batah, Robson Francisco Carvalho and Alexandre Todorovic Fabro; Visualization: José Baddini-Martinez, Vera Luiza Capelozzi, Rosane Duarte Achcar and Alexandre Todorovic Fabro; Writing—original draft: Sabrina Setembre Batah and Alexandre Todorovic Fabro; Writing—review & editing: Sabrina Setembre Batah, Robson Francisco Carvalho, José Baddini-Martinez, Vera Luiza Capelozzi, Suzana Tanni, Rosane Duarte Achcar and Alexandre Todorovic Fabro.
The authors declare no conflict of interest.
This research was supported by São Paulo Research Foundation (Fapesp 22/02821-0; 20/13370-4; 23/10186-6; 23/04199-8; 21/09024-6; 23/10186-6; 23/04199-8; 23/10184-3) and by the National Council for Scientific and Technological Development (CNPq 310415/2021-7).
The Research Ethics Committee approved this study, and written informed consent was waived (CAAE: 43040920.0.0000.5440, 03737018.6.0000.5440 and 65315822.0.0000.5411).
期刊介绍:
Clinical and Translational Medicine (CTM) is an international, peer-reviewed, open-access journal dedicated to accelerating the translation of preclinical research into clinical applications and fostering communication between basic and clinical scientists. It highlights the clinical potential and application of various fields including biotechnologies, biomaterials, bioengineering, biomarkers, molecular medicine, omics science, bioinformatics, immunology, molecular imaging, drug discovery, regulation, and health policy. With a focus on the bench-to-bedside approach, CTM prioritizes studies and clinical observations that generate hypotheses relevant to patients and diseases, guiding investigations in cellular and molecular medicine. The journal encourages submissions from clinicians, researchers, policymakers, and industry professionals.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


