Pharmacological characterization of prostaglandin E2 EP2 and EP4 receptors in male rat locus coeruleus neurons ex vivo
IF 5.6
2区 医学
Q1 PHARMACOLOGY & PHARMACY
引用次数: 0
Abstract
The inflammatory mediator prostaglandin E2 (PGE2) binds to Gs-coupled EP2 and EP4 receptors. These receptors are located in the locus coeruleus (LC), the principal noradrenergic nucleus in the brain, but their functional role remains unknown. In this study, the PGE2 EP2 and EP4 receptors in LC cells from male rat brain slices were pharmacologically characterized by single-unit extracellular electrophysiology. The EP2 receptor agonists butaprost (0.01–10 μM) and treprostinil (0.03–10 µM) and the EP4 receptor agonists rivenprost (0.01 nM–1 µM) and TCS2510 (0.20 nM–2 µM) increased the firing rate of LC neurons in a concentration-dependent manner. The EP2 receptor antagonist PF-04418948 (10 nM) hindered the excitatory effect of butaprost and treprostinil while the EP4 receptor antagonist L-161,982 (30 and 300 nM) blocked the excitatory effect caused by rivenprost and TCS2510. The effects of butaprost and rivenprost were prevented by extracellular sodium replacement but were not modified by the protein kinase A (PKA) activator 8-Br-cAMP (1 mM) or the inhibitor H-89 (10 μM). However, the Gβγ subunit blocker gallein (20 μM) hindered the stimulatory effect of butaprost while the Gαs subunit inhibitor NF449 (10 µM) prevented that of rivenprost. Finally, rivenprost-induced stimulation (30 nM) was not occluded by butaprost (1 µM).
In conclusion, activation of EP2 and EP4 receptors excites LC noradrenergic neurons through sodium-dependent currents via different G protein subunits in male rat brain slices. EP2 and EP4 in the LC may constitute pharmacological targets for the treatment of pain, fever, drug addiction, anxiety and neuroinflammatory diseases.
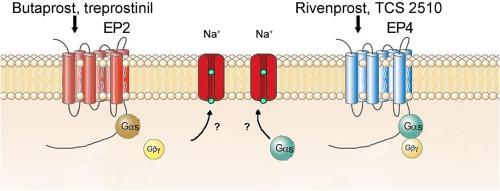
雄性大鼠体外脑室神经元中前列腺素 E2 EP2 和 EP4 受体的药理学特征。
炎症介质前列腺素 E2(PGE2)与 Gs 偶联的 EP2 和 EP4 受体结合。这些受体位于大脑中主要的去甲肾上腺素能神经核(LC),但其功能作用仍不清楚。本研究通过单体胞外电生理学对雄性大鼠脑切片 LC 细胞中的 PGE2 EP2 和 EP4 受体进行了药理学表征。EP2 受体激动剂丁前列素(0.01-10 μM)和曲普前列素(0.03-10 µM)以及 EP4 受体激动剂瑞文前列素(0.01 nM-1 µM)和 TCS2510(0.20 nM-2 µM)以浓度依赖的方式增加了 LC 神经元的发射率。EP2受体拮抗剂PF-04418948(10 nM)阻碍了丁前列素和曲前列素的兴奋作用,而EP4受体拮抗剂L-161982(30 nM和300 nM)阻断了里文前列素和TCS2510引起的兴奋作用。细胞外钠置换可阻止丁前列素和瑞文前列素的作用,但蛋白激酶 A(PKA)激活剂 8-Br-cAMP(1 mM)或抑制剂 H-89(10 μM)不会改变丁前列素和瑞文前列素的作用。然而,Gβγ亚基阻断剂加林(20 μM)阻碍了丁前列素的刺激作用,而Gαs亚基抑制剂NF449(10 μM)则阻止了利文前列素的刺激作用。最后,丁前列素(1 µM)不能阻止里文前列素诱导的刺激(30 nM)。总之,在雄性大鼠脑片中,EP2 和 EP4 受体通过不同的 G 蛋白亚基激活钠依赖性电流,从而兴奋 LC 去甲肾上腺素能神经元。LC中的EP2和EP4可能是治疗疼痛、发热、药物成瘾、焦虑和神经炎症性疾病的药理学靶点。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Biochemical pharmacology
医学-药学
CiteScore
10.30
自引率
1.70%
发文量
420
审稿时长
17 days
期刊介绍:
Biochemical Pharmacology publishes original research findings, Commentaries and review articles related to the elucidation of cellular and tissue function(s) at the biochemical and molecular levels, the modification of cellular phenotype(s) by genetic, transcriptional/translational or drug/compound-induced modifications, as well as the pharmacodynamics and pharmacokinetics of xenobiotics and drugs, the latter including both small molecules and biologics.
The journal''s target audience includes scientists engaged in the identification and study of the mechanisms of action of xenobiotics, biologics and drugs and in the drug discovery and development process.
All areas of cellular biology and cellular, tissue/organ and whole animal pharmacology fall within the scope of the journal. Drug classes covered include anti-infectives, anti-inflammatory agents, chemotherapeutics, cardiovascular, endocrinological, immunological, metabolic, neurological and psychiatric drugs, as well as research on drug metabolism and kinetics. While medicinal chemistry is a topic of complimentary interest, manuscripts in this area must contain sufficient biological data to characterize pharmacologically the compounds reported. Submissions describing work focused predominately on chemical synthesis and molecular modeling will not be considered for review.
While particular emphasis is placed on reporting the results of molecular and biochemical studies, research involving the use of tissue and animal models of human pathophysiology and toxicology is of interest to the extent that it helps define drug mechanisms of action, safety and efficacy.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


