Promoting Water Activation via Molecular Engineering Enables Efficient Asymmetric C–C Coupling during CO2 Electroreduction
IF 14.4
1区 化学
Q1 CHEMISTRY, MULTIDISCIPLINARY
引用次数: 0
Abstract
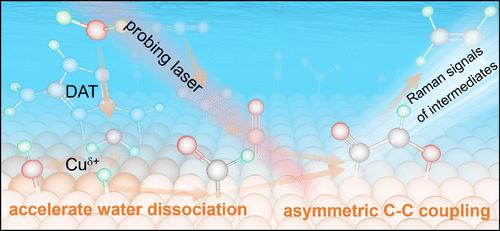
Water activation plays a crucial role in CO2 reduction, but improving the electrocatalytic performance through controlled water activation presents a significant challenge. Herein, we achieved electrochemical CO2 reduction to ethene and ethanol with high selectivity by promoting water dissociation and asymmetric C–C coupling by engineering Cu surfaces with N–H-rich molecules. Direct spectroscopic evidence, coupled with density functional theory calculations, demonstrates that the N–H-rich molecules accelerate interfacial water dissociation via hydrogen-bond interactions, and the generated hydrogen species facilitate the conversion of *CO to *CHO. This enables the efficient asymmetric *CHO–*CO coupling to C2 products with a faradaic efficiency (FE) ∼ 30% higher than that of the unmodified catalyst. Moreover, by adjustment of the relative *CHO/*CO coverage via Cu surface facet regulation, the selectivity can be entirely switched between C2 products and CH4. These mechanistic insights further guided the development of a more efficient catalyst by directly modifying Cu2O nanocubes with the N–H-rich molecule, achieving remarkable C2 product (mainly ethene and ethanol) FEs of 85.7% at a current density of 800 mA cm–2 and excellent stability under nearing industrial conditions. This study advances our understanding of the CO2 reduction mechanisms and offers an effective and general strategy for enhancing electrocatalytic performance by accelerating water dissociation.
通过分子工程促进水活化,在二氧化碳电还原过程中实现高效的不对称 C-C 偶联
水活化在二氧化碳还原过程中起着至关重要的作用,但通过控制水活化来提高电催化性能是一项重大挑战。在此,我们通过在铜表面添加富含 N-H 的分子来促进水的解离和不对称 C-C 偶联,从而实现了高选择性的电化学二氧化碳还原乙烯和乙醇。直接光谱证据以及密度泛函理论计算表明,富含 N-H 的分子通过氢键相互作用加速了界面水解离,生成的氢物种促进了 *CO 向 *CHO 的转化。这使得 *CHO-*CO 与 C2 产物的高效不对称偶联成为可能,其法拉第效率(FE)比未改性催化剂高出 30%。此外,通过调节铜表面的相对 *CHO/*CO 覆盖率,还可以在 C2 产物和 CH4 之间完全切换选择性。这些机理认识进一步指导了通过直接用富 N-H 分子修饰 Cu2O 纳米立方体来开发更高效催化剂的工作,在电流密度为 800 mA cm-2 时,C2 产物(主要是乙烯和乙醇)的显著 FE 为 85.7%,并且在接近工业化的条件下具有极佳的稳定性。这项研究加深了我们对二氧化碳还原机制的理解,并为通过加速水解离来提高电催化性能提供了一种有效的通用策略。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

CiteScore
24.40
自引率
6.00%
发文量
2398
审稿时长
1.6 months
期刊介绍:
The flagship journal of the American Chemical Society, known as the Journal of the American Chemical Society (JACS), has been a prestigious publication since its establishment in 1879. It holds a preeminent position in the field of chemistry and related interdisciplinary sciences. JACS is committed to disseminating cutting-edge research papers, covering a wide range of topics, and encompasses approximately 19,000 pages of Articles, Communications, and Perspectives annually. With a weekly publication frequency, JACS plays a vital role in advancing the field of chemistry by providing essential research.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


