Design, synthesis and therapeutic evaluation of novel antimalarial derivatives based on the clinical antitumor candidate drug Quisinostat
IF 3.3
3区 医学
Q2 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
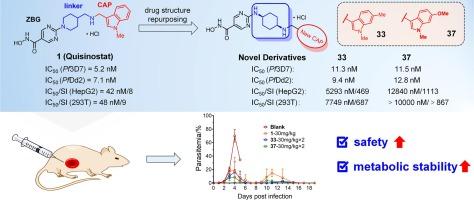
In previous studies, we identified the clinical antitumor drug candidate Quisinostat is a potent Plasmodium falciparum histone deacetylase (PfHDAC) inhibitor with significant activity against drug-resistant malaria but with severe toxicity. To delve deeper into its antimalarial potential, herein we designed and synthesized 36 novel analogues of Quisinostat and systematically evaluated their antimalarial activities and cytotoxicity. Among them, compounds 33 and 37 could effectively eliminate both wild-type and multidrug resistant P. falciparum parasites along with significantly attenuated cytotoxicity, and their metabolic properties were also notably improved. Western blot analysis showed that 33 and 37 upregulated Plasmodium histone acetylation, suggesting that they exerted antimalarial effects through inhibition of PfHDAC like Quisinostat. Furthermore, compounds 33 and 37 also displayed significant antimalarial therapeutic effect and improved animal safety in rodent malaria model. Collectively, 33 and 37 were structurally novel PfHDAC inhibitors and promising antimalarial lead compounds for the next generation of antimalarial drug research.
基于临床抗肿瘤候选药物 Quisinostat 的新型抗疟衍生物的设计、合成和治疗评估
在之前的研究中,我们发现临床抗肿瘤候选药物喹司他是一种强效的恶性疟原虫组蛋白去乙酰化酶(PfHDAC)抑制剂,对抗药性疟疾有显著活性,但毒性严重。为了深入研究其抗疟潜力,我们设计并合成了 36 种新型喹司他类似物,并系统地评估了它们的抗疟活性和细胞毒性。其中,化合物 33 和 37 能有效消灭野生型和耐多药恶性疟原虫寄生虫,细胞毒性显著降低,代谢特性也得到明显改善。Western 印迹分析表明,33 和 37 上调了疟原虫组蛋白乙酰化,表明它们与 Quisinostat 一样通过抑制 PfHDAC 发挥抗疟作用。此外,33 和 37 化合物在啮齿动物疟疾模型中也显示出显著的抗疟疗效,并提高了动物的安全性。总之,33 和 37 是结构新颖的 PfHDAC 抑制剂,是有望用于下一代抗疟药物研究的抗疟先导化合物。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Bioorganic & Medicinal Chemistry
医学-生化与分子生物学
CiteScore
6.80
自引率
2.90%
发文量
413
审稿时长
17 days
期刊介绍:
Bioorganic & Medicinal Chemistry provides an international forum for the publication of full original research papers and critical reviews on molecular interactions in key biological targets such as receptors, channels, enzymes, nucleotides, lipids and saccharides.
The aim of the journal is to promote a better understanding at the molecular level of life processes, and living organisms, as well as the interaction of these with chemical agents. A special feature will be that colour illustrations will be reproduced at no charge to the author, provided that the Editor agrees that colour is essential to the information content of the illustration in question.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


