Sandrine Girard, Magali Pettazzoni, Roseline Froissart, Cécile Pagan, Thomas Boyer, Stephanie Dulucq, Valérie Gonçalves Monteiro, Nicolas Lechevalier, Marie Loosveld, Camille Lours, Caroline Mayeur-Rousse, Mélanie Pannetier, Caroline Peillon, Maria-Alessandra Rosenthal, Sonnthida Sep Hieng, Catherine Trichet, Lucile Baseggio, on behalf the French-Speaking Cellular Haematology Group (GFHC)
{"title":"How to diagnose acid sphingomyelinase deficiency (ASMD) and Niemann–Pick disease type C from bone marrow and peripheral blood smears","authors":"Sandrine Girard, Magali Pettazzoni, Roseline Froissart, Cécile Pagan, Thomas Boyer, Stephanie Dulucq, Valérie Gonçalves Monteiro, Nicolas Lechevalier, Marie Loosveld, Camille Lours, Caroline Mayeur-Rousse, Mélanie Pannetier, Caroline Peillon, Maria-Alessandra Rosenthal, Sonnthida Sep Hieng, Catherine Trichet, Lucile Baseggio, on behalf the French-Speaking Cellular Haematology Group (GFHC)","doi":"10.1002/hem3.70042","DOIUrl":null,"url":null,"abstract":"<p>Lysosomal storage diseases (LSD) are inborn errors of metabolism disorders characterized by a defect in a lysosomal enzyme, transporter, or cofactor. Niemann–Pick diseases are classified into two distinct disorders: acid sphingomyelinase deficiency (ASMD) historically known as Niemann–Pick disease types A, AB, and B, and Niemann-Pick disease type C (NPC).<span><sup>1</sup></span> ASMD is a rare autosomal recessive LSD, caused by pathogenic variants in the ASM-encoding <i>SMPD1</i> gene (OMIM#607608).<span><sup>2</sup></span> It results in the accumulation of sphingomyelin and other lipids, primarily in the liver, spleen, lung, bone marrow, lymph nodes, and central nervous system.<span><sup>3</sup></span> Depending on their clinical phenotype, three different subtypes have been reported: A (severe infantile neurovisceral form), AB (chronic neurovisceral form), and B (chronic visceral form), with a continuum spectrum.<span><sup>1-4</sup></span> NPC is an autosomal recessive LSD caused by the defective function of one of two proteins, NPC1 or NPC2. It results from mutations in the corresponding genes (OMIM#257220 and OMIM#607625). These two proteins act in sequence to regulate the egress of endocytosed nonesterified cholesterol from the late endosomal/lysosomal compartment. NPC manifests as a neurovisceral disease with a highly heterogeneous clinical spectrum.<span><sup>5</sup></span> The prognosis of NPC is correlated with the age of onset of neurological symptoms, with four neurological forms defined: early infantile, late infantile, juvenile, and adolescent-adult. The diagnosis of ASMD and NPC is difficult because these diseases are heterogeneous and may share clinical features with other LSD such as Gaucher disease, especially when splenomegaly is present.<span><sup>5</sup></span> Some cytological abnormalities have been reported in bone marrow (BM) and peripheral blood (PB) smears from ASMD and NPC patients, which could help to guide the more specific analysis such as enzymatic activity, biomarkers measurement, and genetic testing.<span><sup>6</sup></span> However, the cytological data available in the literature are rather limited, often described in single case reports, and do not distinguish the different forms of ASMD and NPC. This work aims to report the cytological features of BM and PB in a retrospective study of 30 French cases from 28 families with ASMD types A [<i>n</i> = 5], AB [<i>n</i> = 3], B [<i>n</i> = 16], and NPC [<i>n</i> = 6], to improve knowledge and define recommendations to assist in diagnosis.</p><p>The diagnosis of cases was based on biochemical analysis, specifically either a deficiency in acid sphingomyelinase activity in blood and/or an abnormal plasma biomarkers profile (i.e., lysosphingomyelin, lysosphingomyelin509/<i>N</i>-palmitoyl-O-phosphocholineserine, and oxysterols), confirmed by specific gene analysis, except for two suspected NPC patients for whom the genetic study was inconclusive and identified a variant of uncertain significance.<span><sup>7</sup></span> Of the 30 cases, 18 were analyzed at diagnosis (10 BM with corresponding PB, 4 BM only, and 4 PB only) and 12 during follow-up, including 9 treated patients (11 PB and 1 BM with corresponding PB). The median age at diagnosis was 8 months (range 6.0–13.0) for ASMD A, 18.0 years for ASMD AB, 53 years (range 20–87.0 months) for ASMD B, and 11.0 years (range 2.0–17.0) for NPC. The male-to-female ratios is 2:3, 1:0, 8:1, and 3:0, respectively.</p><p>Two experienced cytologists (SG, LB) reviewed the cytological features of BM and PB smears following staining with May Grünwald-Giemsa under double-blind conditions using a light microscope. They assessed leukocyte and histiocyte/macrophage characteristics as well as the pattern of the hematopoietic tissue.</p><p>Examination of 14 diagnostic BM smears (Table 1A and Figure 1A) revealed numerous foamy histiocytes (FH) in all cases (ASMD and NPC) often clustered and sometimes isolated. Their nuclei were round or oval, occasionally eccentric, and rarely bi-nucleated. The cytoplasm was abundant, containing numerous small white vacuoles of various sizes, which were highlighted in blue or pink. In some cases, emperipolesis and histiocytes with iron deposits or debris (resembling small black or dark blue beads) were observed. Seven cases exhibited sea-blue histiocytes (SBH), predominantly ASMD B and one in ASMD AB (Figure 1Aa). Vacuolization of hematopoietic cells was observed in three cases, including one case in which vacuolated lymphocytes (VL) were also present in PB (Figure 1Ab). In six cases, adipose vacuoles were more numerous in the spreads, or fatty inclusions were observed in the hematopoietic tissue (Figure 1Ac). Qualitative abnormalities such as dysmyelopoiesis were not noted.</p><p>Examination of 26 PB smears (14 at diagnosis and 12 at follow-up) (Tables 1A,B and Figure 1B) revealed six diagnostic cases with VL exceeding the 5% positivity threshold recommended by the GFHC (5 ASMD A and 1 NPC).<span><sup>6</sup></span> VL exhibited a small number of empty vacuoles (ranging from 2 to 10) with regular contours, often clustered at one pole of the cell (morula-like) or aligned in a manner reminiscent of pearls on a string; they were rarely found in isolation (Figure 1B). In two cases, associated BM smears were analyzed, and only one showed vacuolated cells, including lymphocytes (Figure 1A case 5). No VL was identified in less severe forms at diagnosis (ASMD AB, B and later-onset NPC) or in patients followed at a distance from diagnosis (ASMD AB under enzyme replacement therapy (ERT) [<i>n</i> = 2 siblings], ASMD B ERT treated [<i>n</i> = 4] or untreated [<i>n</i> = 3], and NPC treated [<i>n</i> = 3]).</p><p>When splenomegaly and/or cytopenia are detected, a BM examination is often scheduled. Cytological analysis of BM and PB smears provides valuable information for diagnosing hematological disorders and aids in identifying certain rare LSD. Abnormal accumulation of metabolic by-products within lysosomes may result in the observation of storage cells (large histiocytic storage cells or VL) in BM and PB smears, as well as in prenatal effusions.<span><sup>7</sup></span> Identifying these storage cells can be challenging for untrained observers, as they may resemble storage cells from other LSD or appear similar to reactive cells.</p><p>In BM samples, the primary lesion was characterized by variable proportions of FH and, occasionally, SBH, as described in the literature.<span><sup>1, 9</sup></span> Despite the limited number of cases studied, SBH were primarily identified in patients with ASMD B and one patient with ASMD AB (Table 1A patient 10). SBH were not exclusive and were consistently found in conjunction with non-blue FH. This distinctive cytological appearance is also observed in various other LSD, including gangliosides, ceroid lipofuscinosis, and acquired diseases such as hemoglobinopathies (thalassemia, sickle cell anemia), neoplasms (chronic myeloid leukemia, lymphoma), immunological disorders (idiopathic thrombocytopenic purpura, chronic septic granulomatosis), or lipid storage disorders (SBH syndrome, parenteral nutrition). Interestingly, in patients 2 and 3, the presence of FH guided the diagnosis of LSD in the context of splenomegaly. In these two cases, the NPC biomarker profile was abnormal, but genetic testing did not confirm the diagnosis of NPC. Both patients were found to be compound heterozygous for a pathogenic or likely pathogenic variant and a variant of uncertain significance in the <i>NPC1</i> gene. The clinical significance of variants <i>NPC1</i>:p.Asn222Ser and p.Ser1004Leu is currently debated in the literature and among experts (see ClinVar database). However, these two cases illustrate that such variants, when combined with a pathogenic or likely pathogenic variant, may be associated with hematological manifestations. The densities of SBH and FH were variable and did not correlate with age; this variation appeared to be more related to the cell density of the smear.</p><p>Moreover, analysis of the BM smears revealed that approximately 50% of them exhibited a high lipid content, accompanied by a multitude of adipose vacuoles. No correlation was identified between the patient's lipid profile (when available), age, or disease type, and the proportion of lipid-laden histiocytes. This aspect was more closely related to the quality of the slides, as those that were diluted were less lipid-rich. Cytopenias, particularly thrombocytopenia, are common in patients with ASMD or NPC.<span><sup>1, 5, 10</sup></span> A review of complete blood count results revealed no correlation between the depth or presence of cytopenias and the proportion of BM infiltration by storage histiocytes.</p><p>In PB, VL is not often described in ASMD patients, contrary to NPC or other LSD.<span><sup>11</sup></span> Interestingly, only neurological forms of ASMD (type A) and NPC presented VL in our series. To our knowledge, no cases of ASMD B with VL in PB have been reported in the literature. Distinguishing these VL from reactive lymphocytes present in PB during an infectious episode is crucial.<span><sup>12</sup></span> The latter also display cytoplasmic microvacuoles, which can be observed as a small number of dispersed, fine vacuoles (ranging from one to three in number), or as a multitude of very fine vacuoles, irregularly distributed throughout the cytoplasm, with no discernible order or structure.</p><p>In conclusion, an increased number of FH with white and/or pale blue inclusions is a feature of BM smears in all types of ASMD/NPC. The detection of these FH (which is not limited to SBH alone) represents a significant advancement in the cytological diagnostic approach to ASMD/NPC. The identification of SBH strongly suggests an ASMD type B diagnosis, which requires confirmation by enzymatic activity measurement in combination with <i>SMPD1</i> gene analysis. In contrast, the presence of circulating VL in both PB and BM is uncommon in ASMD and NPC cases. Their presence should prompt consideration of a LSD, particularly a severe form with neurological involvement such as ASMD A or infantile NPC forms. This study represents the largest case series to date, and the results are noteworthy. Further investigation is required in a large prospective study. Finally, a meticulous examination of BM and PB smears is an essential tool to assist in the diagnosis of ASMD and NPC. This analysis is a valuable tool that can help define the different types of ASMD, in addition to the clinical examination and molecular analysis.</p><p>Sandrine Girard and Lucile Baseggio were involved in conceptualization, methodology, collecting data, analysis data, and writing of the original draft preparation. Sandrine Girard, Thomas Boyer, Stephanie Dulucq, Valérie Gonçalves Monteiro, Nicolas Lechevalier, Marie Loosveld, Camille Lours, Caroline Mayeur-Rousse, Mélanie Pannetier, Caroline Peillon, Maria-Alessandra Rosenthal, Sonnthida Sep Hieng, Catherine Trichet provided cases. Sandrine Girard, Mélanie Pannetier, Roseline Froissart, Cécile Pagan, Thomas Boyer, Stephanie Dulucq, Valérie Gonçalves Monteiro, Nicolas Lechevalier, Marie Loosveld, Camille Lours, Caroline Mayeur-Rousse, Mélanie Pannetier, Caroline Peillon, Maria-Alessandra Rosenthal, Sonnthida Sep Hieng, Lucile Baseggio were involved in reviewing the manuscript.</p><p>The authors declare no conflict of interest.</p><p>The study was approved by the Biological Resources Center policy of the Hospices Civils de Lyon. The procedures followed were in accordance with the Declaration of Helsinki. The protocol received approval from the institutional review board (DC-24-5447) This manuscript respects the ethical policy of Hospices Civils de Lyon for the treatment of human research participants. The authors did not obtain written informed consent from the patients but the patients did not object to their data being used for research purposes (as required by the ethics policy of Hospices Civils de Lyon).</p><p>This research received no funding.</p>","PeriodicalId":12982,"journal":{"name":"HemaSphere","volume":"8 11","pages":""},"PeriodicalIF":7.6000,"publicationDate":"2024-11-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11538321/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"HemaSphere","FirstCategoryId":"3","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/hem3.70042","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"HEMATOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Lysosomal storage diseases (LSD) are inborn errors of metabolism disorders characterized by a defect in a lysosomal enzyme, transporter, or cofactor. Niemann–Pick diseases are classified into two distinct disorders: acid sphingomyelinase deficiency (ASMD) historically known as Niemann–Pick disease types A, AB, and B, and Niemann-Pick disease type C (NPC).1 ASMD is a rare autosomal recessive LSD, caused by pathogenic variants in the ASM-encoding SMPD1 gene (OMIM#607608).2 It results in the accumulation of sphingomyelin and other lipids, primarily in the liver, spleen, lung, bone marrow, lymph nodes, and central nervous system.3 Depending on their clinical phenotype, three different subtypes have been reported: A (severe infantile neurovisceral form), AB (chronic neurovisceral form), and B (chronic visceral form), with a continuum spectrum.1-4 NPC is an autosomal recessive LSD caused by the defective function of one of two proteins, NPC1 or NPC2. It results from mutations in the corresponding genes (OMIM#257220 and OMIM#607625). These two proteins act in sequence to regulate the egress of endocytosed nonesterified cholesterol from the late endosomal/lysosomal compartment. NPC manifests as a neurovisceral disease with a highly heterogeneous clinical spectrum.5 The prognosis of NPC is correlated with the age of onset of neurological symptoms, with four neurological forms defined: early infantile, late infantile, juvenile, and adolescent-adult. The diagnosis of ASMD and NPC is difficult because these diseases are heterogeneous and may share clinical features with other LSD such as Gaucher disease, especially when splenomegaly is present.5 Some cytological abnormalities have been reported in bone marrow (BM) and peripheral blood (PB) smears from ASMD and NPC patients, which could help to guide the more specific analysis such as enzymatic activity, biomarkers measurement, and genetic testing.6 However, the cytological data available in the literature are rather limited, often described in single case reports, and do not distinguish the different forms of ASMD and NPC. This work aims to report the cytological features of BM and PB in a retrospective study of 30 French cases from 28 families with ASMD types A [n = 5], AB [n = 3], B [n = 16], and NPC [n = 6], to improve knowledge and define recommendations to assist in diagnosis.
The diagnosis of cases was based on biochemical analysis, specifically either a deficiency in acid sphingomyelinase activity in blood and/or an abnormal plasma biomarkers profile (i.e., lysosphingomyelin, lysosphingomyelin509/N-palmitoyl-O-phosphocholineserine, and oxysterols), confirmed by specific gene analysis, except for two suspected NPC patients for whom the genetic study was inconclusive and identified a variant of uncertain significance.7 Of the 30 cases, 18 were analyzed at diagnosis (10 BM with corresponding PB, 4 BM only, and 4 PB only) and 12 during follow-up, including 9 treated patients (11 PB and 1 BM with corresponding PB). The median age at diagnosis was 8 months (range 6.0–13.0) for ASMD A, 18.0 years for ASMD AB, 53 years (range 20–87.0 months) for ASMD B, and 11.0 years (range 2.0–17.0) for NPC. The male-to-female ratios is 2:3, 1:0, 8:1, and 3:0, respectively.
Two experienced cytologists (SG, LB) reviewed the cytological features of BM and PB smears following staining with May Grünwald-Giemsa under double-blind conditions using a light microscope. They assessed leukocyte and histiocyte/macrophage characteristics as well as the pattern of the hematopoietic tissue.
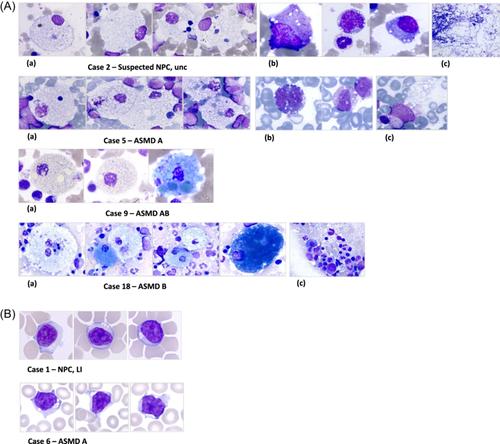
Examination of 14 diagnostic BM smears (Table 1A and Figure 1A) revealed numerous foamy histiocytes (FH) in all cases (ASMD and NPC) often clustered and sometimes isolated. Their nuclei were round or oval, occasionally eccentric, and rarely bi-nucleated. The cytoplasm was abundant, containing numerous small white vacuoles of various sizes, which were highlighted in blue or pink. In some cases, emperipolesis and histiocytes with iron deposits or debris (resembling small black or dark blue beads) were observed. Seven cases exhibited sea-blue histiocytes (SBH), predominantly ASMD B and one in ASMD AB (Figure 1Aa). Vacuolization of hematopoietic cells was observed in three cases, including one case in which vacuolated lymphocytes (VL) were also present in PB (Figure 1Ab). In six cases, adipose vacuoles were more numerous in the spreads, or fatty inclusions were observed in the hematopoietic tissue (Figure 1Ac). Qualitative abnormalities such as dysmyelopoiesis were not noted.
Examination of 26 PB smears (14 at diagnosis and 12 at follow-up) (Tables 1A,B and Figure 1B) revealed six diagnostic cases with VL exceeding the 5% positivity threshold recommended by the GFHC (5 ASMD A and 1 NPC).6 VL exhibited a small number of empty vacuoles (ranging from 2 to 10) with regular contours, often clustered at one pole of the cell (morula-like) or aligned in a manner reminiscent of pearls on a string; they were rarely found in isolation (Figure 1B). In two cases, associated BM smears were analyzed, and only one showed vacuolated cells, including lymphocytes (Figure 1A case 5). No VL was identified in less severe forms at diagnosis (ASMD AB, B and later-onset NPC) or in patients followed at a distance from diagnosis (ASMD AB under enzyme replacement therapy (ERT) [n = 2 siblings], ASMD B ERT treated [n = 4] or untreated [n = 3], and NPC treated [n = 3]).
When splenomegaly and/or cytopenia are detected, a BM examination is often scheduled. Cytological analysis of BM and PB smears provides valuable information for diagnosing hematological disorders and aids in identifying certain rare LSD. Abnormal accumulation of metabolic by-products within lysosomes may result in the observation of storage cells (large histiocytic storage cells or VL) in BM and PB smears, as well as in prenatal effusions.7 Identifying these storage cells can be challenging for untrained observers, as they may resemble storage cells from other LSD or appear similar to reactive cells.
In BM samples, the primary lesion was characterized by variable proportions of FH and, occasionally, SBH, as described in the literature.1, 9 Despite the limited number of cases studied, SBH were primarily identified in patients with ASMD B and one patient with ASMD AB (Table 1A patient 10). SBH were not exclusive and were consistently found in conjunction with non-blue FH. This distinctive cytological appearance is also observed in various other LSD, including gangliosides, ceroid lipofuscinosis, and acquired diseases such as hemoglobinopathies (thalassemia, sickle cell anemia), neoplasms (chronic myeloid leukemia, lymphoma), immunological disorders (idiopathic thrombocytopenic purpura, chronic septic granulomatosis), or lipid storage disorders (SBH syndrome, parenteral nutrition). Interestingly, in patients 2 and 3, the presence of FH guided the diagnosis of LSD in the context of splenomegaly. In these two cases, the NPC biomarker profile was abnormal, but genetic testing did not confirm the diagnosis of NPC. Both patients were found to be compound heterozygous for a pathogenic or likely pathogenic variant and a variant of uncertain significance in the NPC1 gene. The clinical significance of variants NPC1:p.Asn222Ser and p.Ser1004Leu is currently debated in the literature and among experts (see ClinVar database). However, these two cases illustrate that such variants, when combined with a pathogenic or likely pathogenic variant, may be associated with hematological manifestations. The densities of SBH and FH were variable and did not correlate with age; this variation appeared to be more related to the cell density of the smear.
Moreover, analysis of the BM smears revealed that approximately 50% of them exhibited a high lipid content, accompanied by a multitude of adipose vacuoles. No correlation was identified between the patient's lipid profile (when available), age, or disease type, and the proportion of lipid-laden histiocytes. This aspect was more closely related to the quality of the slides, as those that were diluted were less lipid-rich. Cytopenias, particularly thrombocytopenia, are common in patients with ASMD or NPC.1, 5, 10 A review of complete blood count results revealed no correlation between the depth or presence of cytopenias and the proportion of BM infiltration by storage histiocytes.
In PB, VL is not often described in ASMD patients, contrary to NPC or other LSD.11 Interestingly, only neurological forms of ASMD (type A) and NPC presented VL in our series. To our knowledge, no cases of ASMD B with VL in PB have been reported in the literature. Distinguishing these VL from reactive lymphocytes present in PB during an infectious episode is crucial.12 The latter also display cytoplasmic microvacuoles, which can be observed as a small number of dispersed, fine vacuoles (ranging from one to three in number), or as a multitude of very fine vacuoles, irregularly distributed throughout the cytoplasm, with no discernible order or structure.
In conclusion, an increased number of FH with white and/or pale blue inclusions is a feature of BM smears in all types of ASMD/NPC. The detection of these FH (which is not limited to SBH alone) represents a significant advancement in the cytological diagnostic approach to ASMD/NPC. The identification of SBH strongly suggests an ASMD type B diagnosis, which requires confirmation by enzymatic activity measurement in combination with SMPD1 gene analysis. In contrast, the presence of circulating VL in both PB and BM is uncommon in ASMD and NPC cases. Their presence should prompt consideration of a LSD, particularly a severe form with neurological involvement such as ASMD A or infantile NPC forms. This study represents the largest case series to date, and the results are noteworthy. Further investigation is required in a large prospective study. Finally, a meticulous examination of BM and PB smears is an essential tool to assist in the diagnosis of ASMD and NPC. This analysis is a valuable tool that can help define the different types of ASMD, in addition to the clinical examination and molecular analysis.
Sandrine Girard and Lucile Baseggio were involved in conceptualization, methodology, collecting data, analysis data, and writing of the original draft preparation. Sandrine Girard, Thomas Boyer, Stephanie Dulucq, Valérie Gonçalves Monteiro, Nicolas Lechevalier, Marie Loosveld, Camille Lours, Caroline Mayeur-Rousse, Mélanie Pannetier, Caroline Peillon, Maria-Alessandra Rosenthal, Sonnthida Sep Hieng, Catherine Trichet provided cases. Sandrine Girard, Mélanie Pannetier, Roseline Froissart, Cécile Pagan, Thomas Boyer, Stephanie Dulucq, Valérie Gonçalves Monteiro, Nicolas Lechevalier, Marie Loosveld, Camille Lours, Caroline Mayeur-Rousse, Mélanie Pannetier, Caroline Peillon, Maria-Alessandra Rosenthal, Sonnthida Sep Hieng, Lucile Baseggio were involved in reviewing the manuscript.
The authors declare no conflict of interest.
The study was approved by the Biological Resources Center policy of the Hospices Civils de Lyon. The procedures followed were in accordance with the Declaration of Helsinki. The protocol received approval from the institutional review board (DC-24-5447) This manuscript respects the ethical policy of Hospices Civils de Lyon for the treatment of human research participants. The authors did not obtain written informed consent from the patients but the patients did not object to their data being used for research purposes (as required by the ethics policy of Hospices Civils de Lyon).
期刊介绍:
HemaSphere, as a publication, is dedicated to disseminating the outcomes of profoundly pertinent basic, translational, and clinical research endeavors within the field of hematology. The journal actively seeks robust studies that unveil novel discoveries with significant ramifications for hematology.
In addition to original research, HemaSphere features review articles and guideline articles that furnish lucid synopses and discussions of emerging developments, along with recommendations for patient care.
Positioned as the foremost resource in hematology, HemaSphere augments its offerings with specialized sections like HemaTopics and HemaPolicy. These segments engender insightful dialogues covering a spectrum of hematology-related topics, including digestible summaries of pivotal articles, updates on new therapies, deliberations on European policy matters, and other noteworthy news items within the field. Steering the course of HemaSphere are Editor in Chief Jan Cools and Deputy Editor in Chief Claire Harrison, alongside the guidance of an esteemed Editorial Board comprising international luminaries in both research and clinical realms, each representing diverse areas of hematologic expertise.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


