Thermal conductivity of WC: Microstructural design driven by first-principles simulations
IF 8.3
1区 材料科学
Q1 MATERIALS SCIENCE, MULTIDISCIPLINARY
引用次数: 0
Abstract
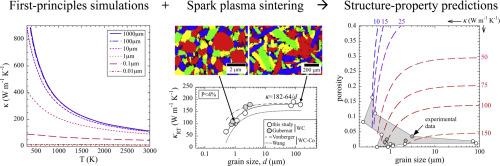
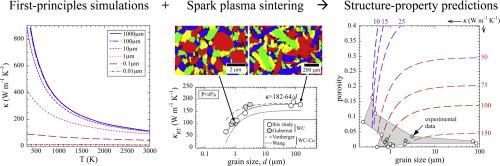
The relationships between the microstructure and the thermal conductivity of binderless WC have been quantified, considering crystal orientation, isotopic abundance, porosity, and grain size. A significantly higher conductivity is predicted in the out-of-plane (c-axis) direction vs. the in-plane (a-axis) direction, using first principles simulations. Isotopic enrichment of the tungsten sublattice is predicted to increase conductivity, e.g., by a factor of 4–5 in the absence of boundary scattering. The results suggest that for an isotopically pure single crystal a thermal conductivity exceeding 1000 W m−1 K−1 may be achievable normal to the basal plane. The conductivity of samples with various porosities could be well fit by a minimum surface area (exponential) model, with a porosity exponent of b = 4.4. Experiment and simulation show a strong grain size dependence to conductivity below 1 µm, with a saturation beyond ∼10 µm. The experimental plateau values for κ were ∼45 % lower than those of the simulations due to deviations from perfect stoichiometry. We also find a higher scattering coefficient in the experiments, likely due to effects of grain size distribution and elongation. Our study clarifies the physical origin of disagreeing literature reports as being predominantly due to grain boundary scattering and enables microstructural design for thermally demanding environments.
碳化钨的导热性:第一原理模拟驱动的微结构设计
考虑到晶体取向、同位素丰度、孔隙率和晶粒尺寸,我们对无粘结剂碳化钨的微观结构与热导率之间的关系进行了量化。根据第一原理模拟预测,面外(c 轴)方向的热导率明显高于面内(a 轴)方向。据预测,钨亚晶格的同位素富集会提高导电率,例如,在没有边界散射的情况下,导电率会提高 4-5 倍。结果表明,对于同位素纯净的单晶体,其热导率可超过 1000 W m-1 K-1。各种孔隙率样品的导热率都可以用最小表面积(指数)模型很好地拟合,孔隙率指数为 b = 4.4。实验和模拟结果表明,1 微米以下的电导率与晶粒大小密切相关,超过 10 微米后达到饱和。由于偏离了完美的化学计量,κ 的实验高原值比模拟值低 45%。我们还发现实验中的散射系数更高,这可能是由于晶粒尺寸分布和伸长率的影响。我们的研究澄清了文献报告中存在分歧的主要原因是晶界散射的物理来源,并使微结构设计能够适应热要求较高的环境。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Acta Materialia
工程技术-材料科学:综合
CiteScore
16.10
自引率
8.50%
发文量
801
审稿时长
53 days
期刊介绍:
Acta Materialia serves as a platform for publishing full-length, original papers and commissioned overviews that contribute to a profound understanding of the correlation between the processing, structure, and properties of inorganic materials. The journal seeks papers with high impact potential or those that significantly propel the field forward. The scope includes the atomic and molecular arrangements, chemical and electronic structures, and microstructure of materials, focusing on their mechanical or functional behavior across all length scales, including nanostructures.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


