Tufm lactylation regulates neuronal apoptosis by modulating mitophagy in traumatic brain injury
IF 13.7
1区 生物学
Q1 BIOCHEMISTRY & MOLECULAR BIOLOGY
引用次数: 0
Abstract
Lactates accumulation following traumatic brain injury (TBI) is detrimental. However, whether lactylation is triggered and involved in the deterioration of TBI remains unknown. Here, we first report that Tufm lactylation pathway induces neuronal apoptosis in TBI. Lactylation is found significantly increased in brain tissues from patients with TBI and mice with controlled cortical impact (CCI), and in neuronal injury cell models. Tufm, a key factor in mitophagy, is screened and identified to be mostly lactylated. Tufm is detected to be lactylated at K286 and the lactylation inhibits the interaction of Tufm and Tomm40 on mitochondria. The mitochondrial distribution of Tufm is then inhibited. Consequently, Tufm-mediated mitophagy is suppressed while mitochondria-induced neuronal apoptosis is increased. In contrast, the knockin of a lactylation-deficient TufmK286R mutant in mice rescues the mitochondrial distribution of Tufm and Tufm-mediated mitophagy, and improves functional outcome after CCI. Likewise, mild hypothermia, as a critical therapeutic method in neuroprotection, helps in downregulating Tufm lactylation, increasing Tufm-mediated mitophagy, mitigating neuronal apoptosis, and eventually ameliorating the outcome of TBI. A novel molecular mechanism in neuronal apoptosis, TBI-initiated Tufm lactylation suppressing mitophagy, is thus revealed.

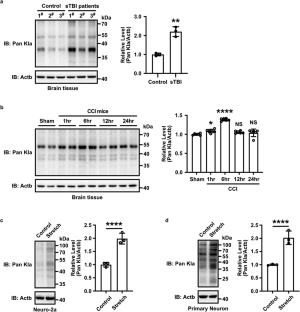
Tufm 乳酰化通过调节创伤性脑损伤中的有丝分裂来调节神经细胞凋亡。
创伤性脑损伤(TBI)后乳酸堆积是有害的。然而,乳酸化是否会引发并参与创伤性脑损伤的恶化仍是未知数。在此,我们首次报道了 Tufm 乳酸化途径在创伤性脑损伤中诱导神经元凋亡。在 TBI 患者和受控皮质冲击(CCI)小鼠的脑组织中,以及在神经元损伤细胞模型中,发现乳化作用明显增加。Tufm 是有丝分裂过程中的一个关键因子,经筛选和鉴定,Tufm 大部分被乳化。检测到 Tufm 在 K286 处被乳化,乳化抑制了线粒体上 Tufm 和 Tomm40 的相互作用。Tufm 的线粒体分布随之受到抑制。因此,Tufm 介导的有丝分裂受到抑制,而线粒体诱导的神经细胞凋亡增加。相反,在小鼠体内敲入乳酸化缺陷的 TufmK286R 突变体可挽救 Tufm 的线粒体分布和 Tufm 介导的有丝分裂,并改善 CCI 后的功能预后。同样,轻度低温作为神经保护的重要治疗方法,有助于下调 Tufm 乳化,增加 Tufm 介导的有丝分裂,减轻神经元凋亡,最终改善创伤性脑损伤的结果。由此揭示了创伤性脑损伤引发的 Tufm 乳化抑制有丝分裂这一神经元凋亡的新分子机制。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Cell Death and Differentiation
生物-生化与分子生物学
CiteScore
24.70
自引率
1.60%
发文量
181
审稿时长
3 months
期刊介绍:
Mission, vision and values of Cell Death & Differentiation:
To devote itself to scientific excellence in the field of cell biology, molecular biology, and biochemistry of cell death and disease.
To provide a unified forum for scientists and clinical researchers
It is committed to the rapid publication of high quality original papers relating to these subjects, together with topical, usually solicited, reviews, meeting reports, editorial correspondence and occasional commentaries on controversial and scientifically informative issues.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


