Buzhong Zhang, Meili Zheng, Yuzhou Zhang, Lijun Quan
{"title":"DCMA: faster protein backbone dihedral angle prediction using a dilated convolutional attention-based neural network.","authors":"Buzhong Zhang, Meili Zheng, Yuzhou Zhang, Lijun Quan","doi":"10.3389/fbinf.2024.1477909","DOIUrl":null,"url":null,"abstract":"<p><p>The dihedral angle of the protein backbone can describe the main structure of the protein, which is of great significance for determining the protein structure. Many computational methods have been proposed to predict this critically important protein structure, including deep learning. However, these heavyweight methods require more computational resources, and the training time becomes intolerable. In this article, we introduce a novel lightweight method, named dilated convolution and multi-head attention (DCMA), that predicts protein backbone torsion dihedral angles <math><mrow><mo>(</mo> <mrow><mi>ϕ</mi> <mo>,</mo> <mi>ψ</mi></mrow> <mo>)</mo></mrow> </math> . DCMA is stacked by five layers of two hybrid inception blocks and one multi-head attention block (I2A1) module. The hybrid inception blocks consisting of multi-scale convolutional neural networks and dilated convolutional neural networks are designed for capturing local and long-range sequence-based features. The multi-head attention block supplementally strengthens this operation. The proposed DCMA is validated on public critical assessment of protein structure prediction (CASP) benchmark datasets. Experimental results show that DCMA obtains better or comparable generalization performance. Compared to best-so-far methods, which are mostly ensemble models and constructed of recurrent neural networks, DCMA is an individual model that is more lightweight and has a shorter training time. The proposed model could be applied as an alternative method for predicting other protein structural features.</p>","PeriodicalId":73066,"journal":{"name":"Frontiers in bioinformatics","volume":"4 ","pages":"1477909"},"PeriodicalIF":3.9000,"publicationDate":"2024-10-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11527783/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Frontiers in bioinformatics","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.3389/fbinf.2024.1477909","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q2","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
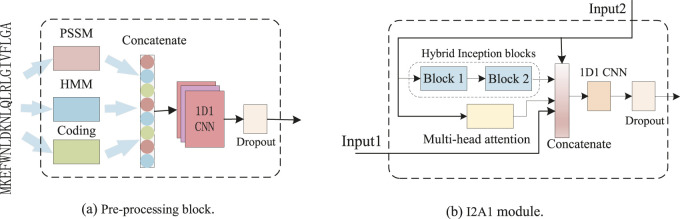
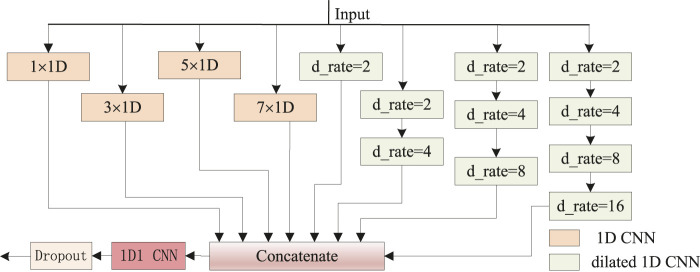
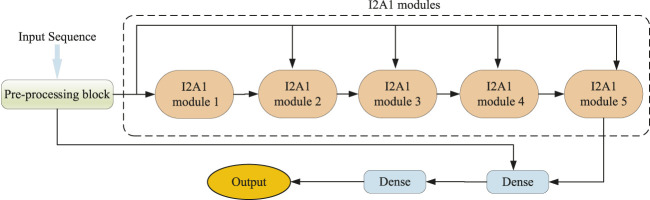
The dihedral angle of the protein backbone can describe the main structure of the protein, which is of great significance for determining the protein structure. Many computational methods have been proposed to predict this critically important protein structure, including deep learning. However, these heavyweight methods require more computational resources, and the training time becomes intolerable. In this article, we introduce a novel lightweight method, named dilated convolution and multi-head attention (DCMA), that predicts protein backbone torsion dihedral angles . DCMA is stacked by five layers of two hybrid inception blocks and one multi-head attention block (I2A1) module. The hybrid inception blocks consisting of multi-scale convolutional neural networks and dilated convolutional neural networks are designed for capturing local and long-range sequence-based features. The multi-head attention block supplementally strengthens this operation. The proposed DCMA is validated on public critical assessment of protein structure prediction (CASP) benchmark datasets. Experimental results show that DCMA obtains better or comparable generalization performance. Compared to best-so-far methods, which are mostly ensemble models and constructed of recurrent neural networks, DCMA is an individual model that is more lightweight and has a shorter training time. The proposed model could be applied as an alternative method for predicting other protein structural features.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


