{"title":"Long-Term Observation of Focal Segmental Glomerulosclerosis after Treatment of Renal Parenchymal Malakoplakia: A Case Report.","authors":"Reiji Takami, Yoshikuni Nagayama, Hiroki Nishiwaki, Toshiharu Ueno, Shigeki Iwasaki, Ashio Yoshimura, Fumihiko Koiwa","doi":"10.1159/000540877","DOIUrl":null,"url":null,"abstract":"<p><strong>Introduction: </strong>Malakoplakia is a rare and chronic granulomatous disease that is pathologically characterized by the presence of Michaelis-Gutmann bodies and large macrophage clusters. Malakoplakia of the renal parenchyma is especially rare. In this report, we describe the long-term prognosis of a patient who was diagnosed with and treated for renal parenchymal malakoplakia in infancy.</p><p><strong>Case presentation: </strong>Seventeen years after malakoplakia onset, the patient presented to us with worsening proteinuria. Computed tomography revealed structural abnormalities in the kidney, and focal segmental glomerulosclerosis (FSGS) was diagnosed based on renal biopsy findings. No Michaelis-Gutmann bodies were observed in von Kossa-stained biopsy specimens. Regular outpatient monitoring during the next 9 years showed gradual deterioration of renal function and a moderately high protein/creatinine ratio.</p><p><strong>Conclusion: </strong>Our findings suggest that structural changes due to malakoplakia can cause FSGS. Moreover, structural changes indicate the healing of malakoplakia in infancy and the disappearance of its characteristic lesions over time. Owing to its long-term observation period, this unique case provides new insights into the outcomes of patients with renal parenchymal malakoplakia.</p>","PeriodicalId":9599,"journal":{"name":"Case Reports in Nephrology and Dialysis","volume":"14 1","pages":"158-163"},"PeriodicalIF":0.9000,"publicationDate":"2024-10-07","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11521469/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Case Reports in Nephrology and Dialysis","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1159/000540877","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/1/1 0:00:00","PubModel":"eCollection","JCR":"Q4","JCRName":"UROLOGY & NEPHROLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Introduction: Malakoplakia is a rare and chronic granulomatous disease that is pathologically characterized by the presence of Michaelis-Gutmann bodies and large macrophage clusters. Malakoplakia of the renal parenchyma is especially rare. In this report, we describe the long-term prognosis of a patient who was diagnosed with and treated for renal parenchymal malakoplakia in infancy.
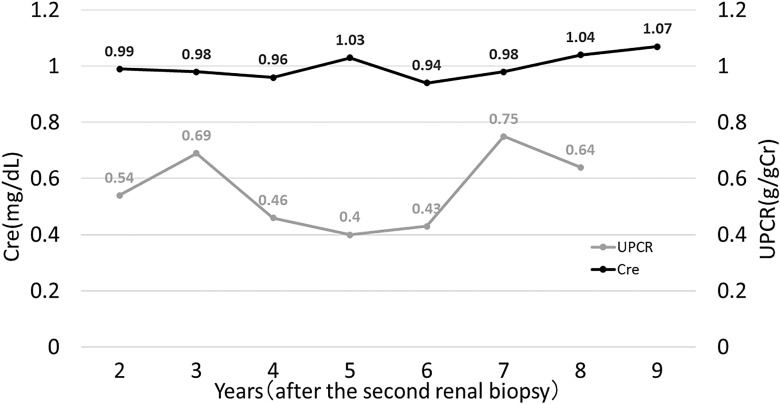
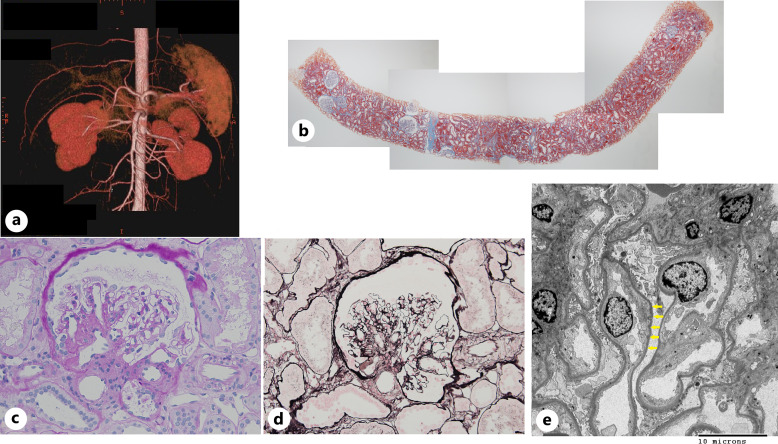
Case presentation: Seventeen years after malakoplakia onset, the patient presented to us with worsening proteinuria. Computed tomography revealed structural abnormalities in the kidney, and focal segmental glomerulosclerosis (FSGS) was diagnosed based on renal biopsy findings. No Michaelis-Gutmann bodies were observed in von Kossa-stained biopsy specimens. Regular outpatient monitoring during the next 9 years showed gradual deterioration of renal function and a moderately high protein/creatinine ratio.
Conclusion: Our findings suggest that structural changes due to malakoplakia can cause FSGS. Moreover, structural changes indicate the healing of malakoplakia in infancy and the disappearance of its characteristic lesions over time. Owing to its long-term observation period, this unique case provides new insights into the outcomes of patients with renal parenchymal malakoplakia.
期刊介绍:
This peer-reviewed online-only journal publishes original case reports covering the entire spectrum of nephrology and dialysis, including genetic susceptibility, clinical presentation, diagnosis, treatment or prevention, toxicities of therapy, critical care, supportive care, quality-of-life and survival issues. The journal will also accept case reports dealing with the use of novel technologies, both in the arena of diagnosis and treatment. Supplementary material is welcomed.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


