Jessica Valero-Rojas, Camilo Ramírez-Sánchez, Laura Pacheco-Paternina, Paulina Valenzuela-Hormazabal, Fernanda I Saldivar-González, Paula Santana, Janneth González, Tatiana Gutiérrez-Bunster, Alejandro Valdés-Jiménez, David Ramírez
{"title":"AlzyFinder: A Machine-Learning-Driven Platform for Ligand-Based Virtual Screening and Network Pharmacology.","authors":"Jessica Valero-Rojas, Camilo Ramírez-Sánchez, Laura Pacheco-Paternina, Paulina Valenzuela-Hormazabal, Fernanda I Saldivar-González, Paula Santana, Janneth González, Tatiana Gutiérrez-Bunster, Alejandro Valdés-Jiménez, David Ramírez","doi":"10.1021/acs.jcim.4c01481","DOIUrl":null,"url":null,"abstract":"<p><p>Alzheimer's disease (AD), a prevalent neurodegenerative disorder, presents significant challenges in drug development due to its multifactorial nature and complex pathophysiology. The AlzyFinder Platform, introduced in this study, addresses these challenges by providing a comprehensive, free web-based tool for parallel ligand-based virtual screening and network pharmacology, specifically targeting over 85 key proteins implicated in AD. This innovative approach is designed to enhance the identification and analysis of potential multitarget ligands, thereby accelerating the development of effective therapeutic strategies against AD. AlzyFinder Platform incorporates machine learning models to facilitate the ligand-based virtual screening process. These models, built with the XGBoost algorithm and optimized through Optuna, were meticulously trained and validated using robust methodologies to ensure high predictive accuracy. Validation included extensive testing with active, inactive, and decoy molecules, demonstrating the platform's efficacy in distinguishing active compounds. The models are evaluated based on balanced accuracy, precision, and F1 score metrics. A unique soft-voting ensemble approach is utilized to refine the classification process, integrating the strengths of individual models. This methodological framework enables a comprehensive analysis of interaction data, which is presented in multiple formats such as tables, heat maps, and interactive Ligand-Protein Interaction networks, thus enhancing the visualization and analysis of drug-protein interactions. AlzyFinder was applied to screen five molecules recently reported (and not used to train or validate the ML models) as active compounds against five key AD targets. The platform demonstrated its efficacy by accurately predicting all five molecules as true positives with a probability greater than 0.70. This result underscores the platform's capability in identifying potential therapeutic compounds with high precision. In conclusion, AlzyFinder's innovative approach extends beyond traditional virtual screening by incorporating network pharmacology analysis, thus providing insights into the systemic actions of drug candidates. This feature allows for the exploration of ligand-protein and protein-protein interactions and their extensions, offering a comprehensive view of potential therapeutic impacts. As the first open-access platform of its kind, AlzyFinder stands as a valuable resource for the AD research community, available at http://www.alzyfinder-platform.udec.cl with supporting data and scripts accessible via GitHub https://github.com/ramirezlab/AlzyFinder.</p>","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":" ","pages":"9040-9047"},"PeriodicalIF":5.6000,"publicationDate":"2024-12-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c01481","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/31 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
引用次数: 0
Abstract
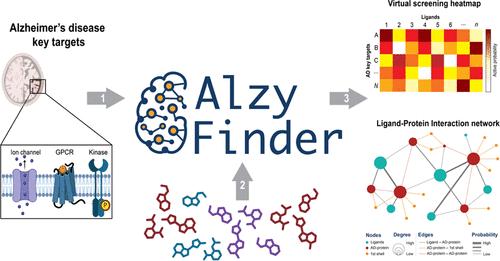
Alzheimer's disease (AD), a prevalent neurodegenerative disorder, presents significant challenges in drug development due to its multifactorial nature and complex pathophysiology. The AlzyFinder Platform, introduced in this study, addresses these challenges by providing a comprehensive, free web-based tool for parallel ligand-based virtual screening and network pharmacology, specifically targeting over 85 key proteins implicated in AD. This innovative approach is designed to enhance the identification and analysis of potential multitarget ligands, thereby accelerating the development of effective therapeutic strategies against AD. AlzyFinder Platform incorporates machine learning models to facilitate the ligand-based virtual screening process. These models, built with the XGBoost algorithm and optimized through Optuna, were meticulously trained and validated using robust methodologies to ensure high predictive accuracy. Validation included extensive testing with active, inactive, and decoy molecules, demonstrating the platform's efficacy in distinguishing active compounds. The models are evaluated based on balanced accuracy, precision, and F1 score metrics. A unique soft-voting ensemble approach is utilized to refine the classification process, integrating the strengths of individual models. This methodological framework enables a comprehensive analysis of interaction data, which is presented in multiple formats such as tables, heat maps, and interactive Ligand-Protein Interaction networks, thus enhancing the visualization and analysis of drug-protein interactions. AlzyFinder was applied to screen five molecules recently reported (and not used to train or validate the ML models) as active compounds against five key AD targets. The platform demonstrated its efficacy by accurately predicting all five molecules as true positives with a probability greater than 0.70. This result underscores the platform's capability in identifying potential therapeutic compounds with high precision. In conclusion, AlzyFinder's innovative approach extends beyond traditional virtual screening by incorporating network pharmacology analysis, thus providing insights into the systemic actions of drug candidates. This feature allows for the exploration of ligand-protein and protein-protein interactions and their extensions, offering a comprehensive view of potential therapeutic impacts. As the first open-access platform of its kind, AlzyFinder stands as a valuable resource for the AD research community, available at http://www.alzyfinder-platform.udec.cl with supporting data and scripts accessible via GitHub https://github.com/ramirezlab/AlzyFinder.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


