Intrinsic reactivity of stoichiometric IrO2(1 1 0) surface toward oxidative coupling of methane
IF 3
3区 化学
Q3 CHEMISTRY, PHYSICAL
引用次数: 0
Abstract
This study investigates the oxidative coupling of methane on the IrO2(1 1 0) surface using TPRS simulations informed by DFT-derived data. We discover that the efficiency of the IrO2(1 1 0) surface in generating ethylene is strongly influenced by methane surface coverage. Our simulations reveal that the presence of surface hydroxyl group enhances the yield of C2+ species from methane oxidation, but this effect is counteracted at high methane coverages due to the accelerated formation of CH2OH. In addition, the study reveals that slight modifications in energy barriers at the branching point (C2H4 formation vs. CH2OH formation) significantly affect C2H4(g) production from the simulation, underscoring the importance of precise energetic data for accurate catalytic reaction predictions. The results have broader implications for reactions and catalysts where branching point selectivity determines high-value product yields. Thus, combining surface science with computational analysis is crucial for accurately determining energy profiles of key steps at the branching points.
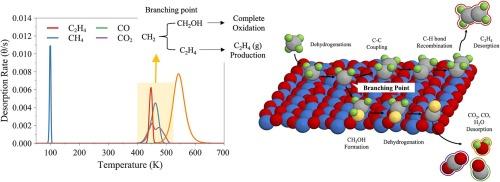
定量IrO2(1 1 0)表面对甲烷氧化偶联的内在反应性
本研究利用 TPRS 模拟和 DFT 衍生数据,研究了甲烷在 IrO2(1 1 0) 表面的氧化耦合。我们发现,IrO2(1 1 0) 表面生成乙烯的效率受到甲烷表面覆盖率的强烈影响。我们的模拟发现,表面羟基的存在提高了甲烷氧化产生的 C2+ 物种的产量,但在甲烷覆盖率较高时,由于 CH2OH 的加速形成,这种效应被抵消。此外,研究还发现,分支点(C2H4 的形成与 CH2OH 的形成)的能量障碍稍有改变,就会显著影响模拟产生的 C2H4(g),这凸显了精确的能量数据对于准确预测催化反应的重要性。这些结果对支化点选择性决定高价值产品产量的反应和催化剂具有更广泛的意义。因此,将表面科学与计算分析相结合对于准确确定分支点关键步骤的能量曲线至关重要。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational and Theoretical Chemistry
CHEMISTRY, PHYSICAL-
CiteScore
4.20
自引率
10.70%
发文量
331
审稿时长
31 days
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


