Alice Lemoinne, Guillaume Dirberg, Myriam Georges, Tony Robinet
{"title":"Evaluation of a Nanopore Sequencing Strategy on Bacterial Communities From Marine Sediments","authors":"Alice Lemoinne, Guillaume Dirberg, Myriam Georges, Tony Robinet","doi":"10.1002/edn3.70009","DOIUrl":null,"url":null,"abstract":"<p>Following the development of high-throughput DNA sequencers, environmental prokaryotic communities were usually described by metabarcoding on short markers of the 16S domain. Among third-generation sequencers, that offered the possibility to sequence the full 16S domain, the portable MinION from Oxford Nanopore was undervalued for metabarcoding because of its relatively higher error rate per read. Here we illustrate the limits and benefits of Nanopore sequencing devices by comparing the prokaryotic community structure in a mock community and 52 sediment samples from mangrove sites, inferred from full-length 16S long-reads (16S-FL, ca. 1.5 kbp) on a MinION device, with those inferred from partial 16S short-reads (16S-V4V5, ca. 0.4 kbp, 16S-V4V5) on Illumina MiSeq. 16S-V4V5 and 16S-FL retrieved all the bacterial species from the mock, but Nanopore long-reads overestimated their diversity (56 species vs. 15). Whether these supplementary OTUs were artefactual or not, they only accounted for ca. 10% of the reads. From the sediment samples, with a coverage-based rarefaction of reads and after singletons filtering, Mantel and Procrustean tests of co-inertia showed that bacterial community structures inferred from 16S-V4V5 and 16S-FL were significantly similar, showing both a comparable contrast between sites and a coherent sea-land orientation within sites. In our dataset, 84.7% and 98.8% of the 16S-V4V5 assigned reads were assigned strictly to the same species and genus, respectively, than those detected by 16S-FL. 16S-FL detected 92.2% of the 309 families and 87.7% of the 448 genera that were detected by the short 16S-V4V5. 16S-FL recorded 973 additional species and 392 genus not detected by 16S-V4V5 (31.5% and 10.4% of the 16S-FL reads, respectively, among which 67.8% and 79.3% were assigned), produced by both primer specificities and different error rates. Thus, our results concluded an overall similarity between 16S-V4V5 and 16S-FL sequencing strategies for this type of environmental samples.</p>","PeriodicalId":52828,"journal":{"name":"Environmental DNA","volume":"6 5","pages":""},"PeriodicalIF":0.0000,"publicationDate":"2024-10-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/edn3.70009","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Environmental DNA","FirstCategoryId":"1085","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/edn3.70009","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"Agricultural and Biological Sciences","Score":null,"Total":0}
引用次数: 0
Abstract
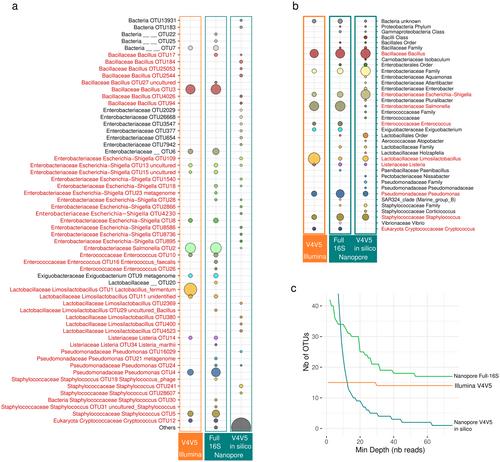
Following the development of high-throughput DNA sequencers, environmental prokaryotic communities were usually described by metabarcoding on short markers of the 16S domain. Among third-generation sequencers, that offered the possibility to sequence the full 16S domain, the portable MinION from Oxford Nanopore was undervalued for metabarcoding because of its relatively higher error rate per read. Here we illustrate the limits and benefits of Nanopore sequencing devices by comparing the prokaryotic community structure in a mock community and 52 sediment samples from mangrove sites, inferred from full-length 16S long-reads (16S-FL, ca. 1.5 kbp) on a MinION device, with those inferred from partial 16S short-reads (16S-V4V5, ca. 0.4 kbp, 16S-V4V5) on Illumina MiSeq. 16S-V4V5 and 16S-FL retrieved all the bacterial species from the mock, but Nanopore long-reads overestimated their diversity (56 species vs. 15). Whether these supplementary OTUs were artefactual or not, they only accounted for ca. 10% of the reads. From the sediment samples, with a coverage-based rarefaction of reads and after singletons filtering, Mantel and Procrustean tests of co-inertia showed that bacterial community structures inferred from 16S-V4V5 and 16S-FL were significantly similar, showing both a comparable contrast between sites and a coherent sea-land orientation within sites. In our dataset, 84.7% and 98.8% of the 16S-V4V5 assigned reads were assigned strictly to the same species and genus, respectively, than those detected by 16S-FL. 16S-FL detected 92.2% of the 309 families and 87.7% of the 448 genera that were detected by the short 16S-V4V5. 16S-FL recorded 973 additional species and 392 genus not detected by 16S-V4V5 (31.5% and 10.4% of the 16S-FL reads, respectively, among which 67.8% and 79.3% were assigned), produced by both primer specificities and different error rates. Thus, our results concluded an overall similarity between 16S-V4V5 and 16S-FL sequencing strategies for this type of environmental samples.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


