Naqash H. Malik, Qaiser Rafiq, Muhammad Farooq Nasir, Sikander Azam, Muhammad Tahir Khan, Gaber A. M. Mersal, Mahmoud M. Hessien
{"title":"Investigating the Optoelectronic Properties of 2-D and 3-D CaTi1−xCuxO3 as a Phosphor Materials: A Density Functional Theory Approach","authors":"Naqash H. Malik, Qaiser Rafiq, Muhammad Farooq Nasir, Sikander Azam, Muhammad Tahir Khan, Gaber A. M. Mersal, Mahmoud M. Hessien","doi":"10.1002/qua.27486","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>The CaTiO<sub>3</sub> has been extensively investigated as a highly promising optical material mostly for its optoelectronic properties and its function as a host for transition metals doped in the CaTiO<sub>3</sub>. Electronic and optical properties of CaTi<sub>1−x</sub>Cu<sub>x</sub>O<sub>3</sub> (2-D and 3-D) have been thoroughly analyzed using first-principles calculations based on Density Functional Theory (DFT). The calculations of these properties in both 2-D and 3-D configurations have performed by the use of generalized gradient approximation plus Hubbard (GGA + U). The electronic characteristics including the electronic band structure, partial density of states, and total density of states have been meticulously computed for CaTi<sub>1−x</sub>Cu<sub>x</sub>O<sub>3</sub> in both 2-D and 3-D. Upon analyzing the obtained results, we investigated that conduction and valence bands overlapped for both 2-D and 3-D structures revealing the metallic nature. We observed transitions mainly attributed to Cu-d, Ti-d, Ti-p, and O-p orbitals in both 2-D and 3-D configurations. Discussion delves into the significance of electronic band structure calculations in understanding optical properties. Peaks in the energy loss function are observed at 13 eV in both cases referred to the plasmon energy. Static values of the dielectric functions, extinction coefficient, reflectivity, and refraction are also computed. Our obtained results showed that the CaTi<sub>1−x</sub>Cu<sub>x</sub>O<sub>3</sub> compound in 3-D form is more apt for optoelectronic devices and UV-LED applications.</p>\n </div>","PeriodicalId":182,"journal":{"name":"International Journal of Quantum Chemistry","volume":"124 21","pages":""},"PeriodicalIF":2.3000,"publicationDate":"2024-10-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"International Journal of Quantum Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/qua.27486","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
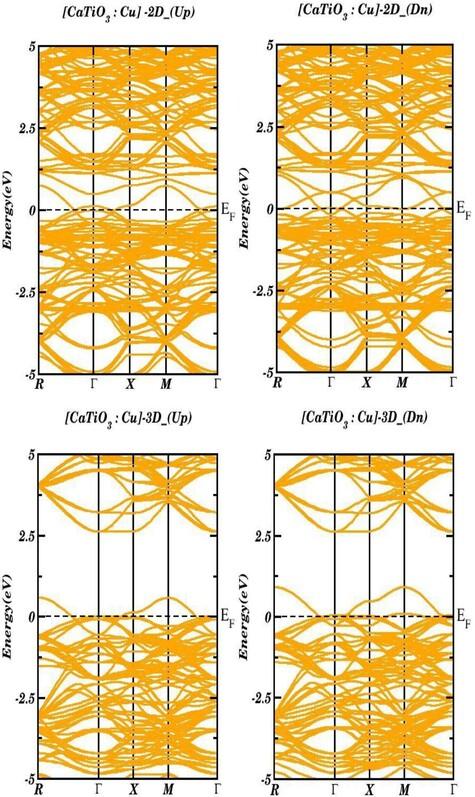
The CaTiO3 has been extensively investigated as a highly promising optical material mostly for its optoelectronic properties and its function as a host for transition metals doped in the CaTiO3. Electronic and optical properties of CaTi1−xCuxO3 (2-D and 3-D) have been thoroughly analyzed using first-principles calculations based on Density Functional Theory (DFT). The calculations of these properties in both 2-D and 3-D configurations have performed by the use of generalized gradient approximation plus Hubbard (GGA + U). The electronic characteristics including the electronic band structure, partial density of states, and total density of states have been meticulously computed for CaTi1−xCuxO3 in both 2-D and 3-D. Upon analyzing the obtained results, we investigated that conduction and valence bands overlapped for both 2-D and 3-D structures revealing the metallic nature. We observed transitions mainly attributed to Cu-d, Ti-d, Ti-p, and O-p orbitals in both 2-D and 3-D configurations. Discussion delves into the significance of electronic band structure calculations in understanding optical properties. Peaks in the energy loss function are observed at 13 eV in both cases referred to the plasmon energy. Static values of the dielectric functions, extinction coefficient, reflectivity, and refraction are also computed. Our obtained results showed that the CaTi1−xCuxO3 compound in 3-D form is more apt for optoelectronic devices and UV-LED applications.
期刊介绍:
Since its first formulation quantum chemistry has provided the conceptual and terminological framework necessary to understand atoms, molecules and the condensed matter. Over the past decades synergistic advances in the methodological developments, software and hardware have transformed quantum chemistry in a truly interdisciplinary science that has expanded beyond its traditional core of molecular sciences to fields as diverse as chemistry and catalysis, biophysics, nanotechnology and material science.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


