Emma McCune, Anukriti Sharma, Breanna Johnson, Tess O'Meara, Sarah Theiner, Maribel Campos, Diane Heditsian, Susie Brain, Jack A Gilbert, Laura Esserman, Michael J Campbell
{"title":"Gut and oral microbial compositional differences in women with breast cancer, women with ductal carcinoma <i>in situ</i>, and healthy women.","authors":"Emma McCune, Anukriti Sharma, Breanna Johnson, Tess O'Meara, Sarah Theiner, Maribel Campos, Diane Heditsian, Susie Brain, Jack A Gilbert, Laura Esserman, Michael J Campbell","doi":"10.1128/msystems.01237-24","DOIUrl":null,"url":null,"abstract":"<p><p>This study characterized and compared the fecal and oral microbiota from women with early-stage breast cancer (BC), women with ductal carcinoma <i>in situ</i> (DCIS), and healthy women. Fecal and oral samples were collected from newly diagnosed patients prior to any therapy and characterized using 16S rRNA sequencing. Measures of gut microbial alpha diversity were significantly lower in the BC vs healthy cohort. Beta diversity differed significantly between the BC or DCIS and healthy groups, and several differentially abundant taxa were identified. Clustering (non-negative matrix factorization) of the gut microbiota identified five bacterial guilds dominated by <i>Prevotella</i>, Enterobacteriaceae, <i>Akkermansia</i>, Clostridiales, or <i>Bacteroides</i>. The <i>Bacteroides</i> and Enterobacteriaceae guilds were significantly more abundant in the BC cohort compared to healthy controls, whereas the Clostridiales guild was more abundant in the healthy group. Finally, prediction of functional pathways identified 23 pathways that differed between the BC and healthy gut microbiota including lipopolysaccharide biosynthesis, glycan biosynthesis and metabolism, lipid metabolism, and sphingolipid metabolism. In contrast to the gut microbiomes, there were no significant differences in alpha or beta diversity in the oral microbiomes, and very few differentially abundant taxa were observed. Non-negative matrix factorization analysis of the oral microbiota samples identified seven guilds dominated by <i>Veillonella</i>, <i>Prevotella</i>, Gemellaceae, <i>Haemophilus</i>, <i>Neisseria</i>, <i>Propionibacterium</i>, and <i>Streptococcus</i>; however, none of these guilds were differentially associated with the different cohorts. Our results suggest that alterations in the gut microbiota may provide the basis for interventions targeting the gut microbiome to improve treatment outcomes and long-term prognosis.</p><p><strong>Importance: </strong>Emerging evidence suggests that the gut microbiota may play a role in breast cancer. Few studies have evaluated both the gut and oral microbiomes in women with breast cancer (BC), and none have characterized these microbiomes in women with ductal carcinoma <i>in situ</i> (DCIS). We surveyed the gut and oral microbiomes from women with BC or DCIS and healthy women and identified compositional and functional features of the gut microbiota that differed between these cohorts. In contrast, very few differential features were identified in the oral microbiota. Understanding the role of gut bacteria in BC and DCIS may open up new opportunities for the development of novel markers for early detection (or markers of susceptibility) as well as new strategies for prevention and/or treatment.</p>","PeriodicalId":18819,"journal":{"name":"mSystems","volume":" ","pages":"e0123724"},"PeriodicalIF":4.6000,"publicationDate":"2024-11-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11575313/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"mSystems","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1128/msystems.01237-24","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/29 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"MICROBIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
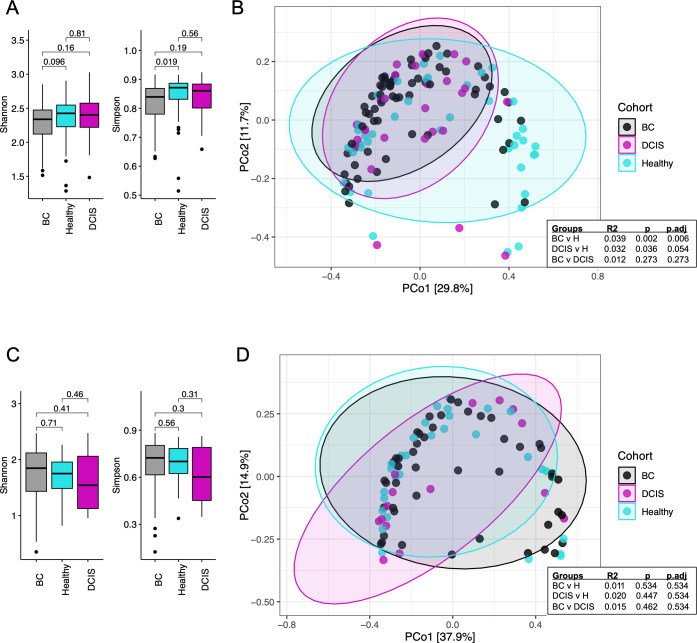
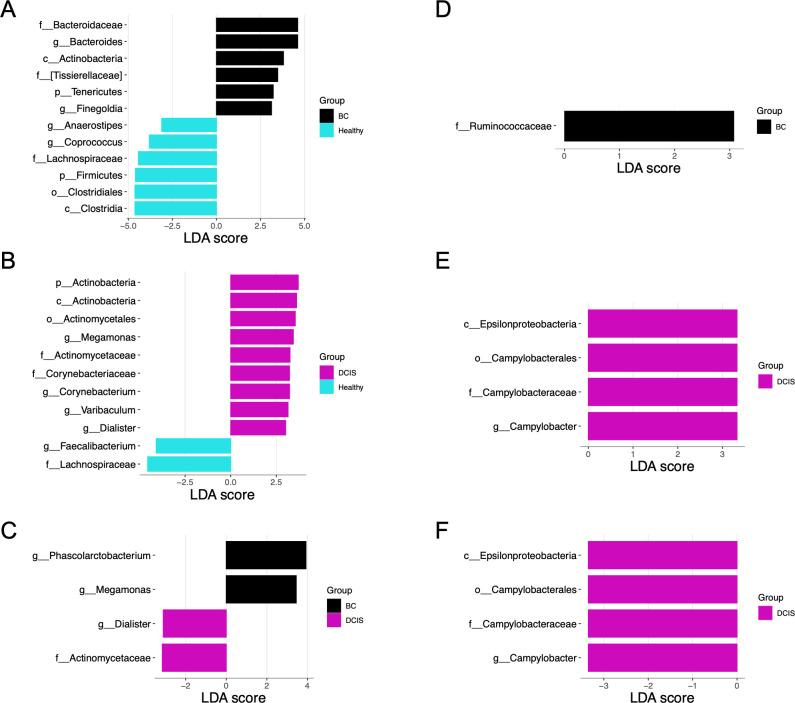
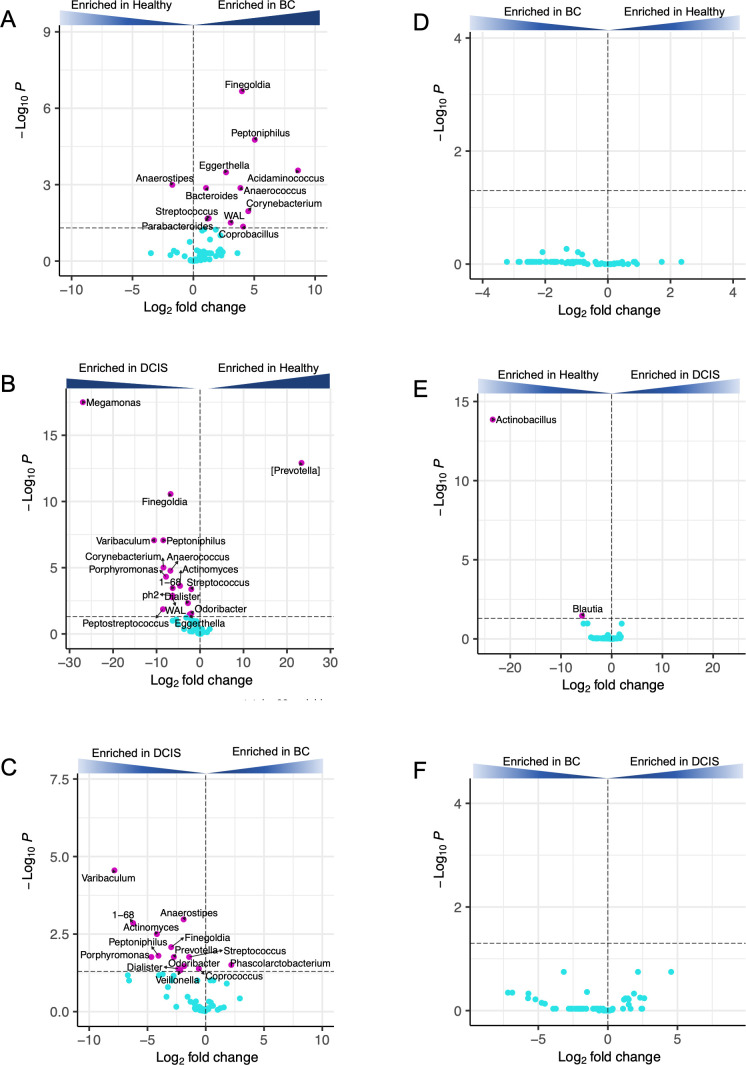
This study characterized and compared the fecal and oral microbiota from women with early-stage breast cancer (BC), women with ductal carcinoma in situ (DCIS), and healthy women. Fecal and oral samples were collected from newly diagnosed patients prior to any therapy and characterized using 16S rRNA sequencing. Measures of gut microbial alpha diversity were significantly lower in the BC vs healthy cohort. Beta diversity differed significantly between the BC or DCIS and healthy groups, and several differentially abundant taxa were identified. Clustering (non-negative matrix factorization) of the gut microbiota identified five bacterial guilds dominated by Prevotella, Enterobacteriaceae, Akkermansia, Clostridiales, or Bacteroides. The Bacteroides and Enterobacteriaceae guilds were significantly more abundant in the BC cohort compared to healthy controls, whereas the Clostridiales guild was more abundant in the healthy group. Finally, prediction of functional pathways identified 23 pathways that differed between the BC and healthy gut microbiota including lipopolysaccharide biosynthesis, glycan biosynthesis and metabolism, lipid metabolism, and sphingolipid metabolism. In contrast to the gut microbiomes, there were no significant differences in alpha or beta diversity in the oral microbiomes, and very few differentially abundant taxa were observed. Non-negative matrix factorization analysis of the oral microbiota samples identified seven guilds dominated by Veillonella, Prevotella, Gemellaceae, Haemophilus, Neisseria, Propionibacterium, and Streptococcus; however, none of these guilds were differentially associated with the different cohorts. Our results suggest that alterations in the gut microbiota may provide the basis for interventions targeting the gut microbiome to improve treatment outcomes and long-term prognosis.
Importance: Emerging evidence suggests that the gut microbiota may play a role in breast cancer. Few studies have evaluated both the gut and oral microbiomes in women with breast cancer (BC), and none have characterized these microbiomes in women with ductal carcinoma in situ (DCIS). We surveyed the gut and oral microbiomes from women with BC or DCIS and healthy women and identified compositional and functional features of the gut microbiota that differed between these cohorts. In contrast, very few differential features were identified in the oral microbiota. Understanding the role of gut bacteria in BC and DCIS may open up new opportunities for the development of novel markers for early detection (or markers of susceptibility) as well as new strategies for prevention and/or treatment.
mSystemsBiochemistry, Genetics and Molecular Biology-Biochemistry
CiteScore
10.50
自引率
3.10%
发文量
308
审稿时长
13 weeks
期刊介绍:
mSystems™ will publish preeminent work that stems from applying technologies for high-throughput analyses to achieve insights into the metabolic and regulatory systems at the scale of both the single cell and microbial communities. The scope of mSystems™ encompasses all important biological and biochemical findings drawn from analyses of large data sets, as well as new computational approaches for deriving these insights. mSystems™ will welcome submissions from researchers who focus on the microbiome, genomics, metagenomics, transcriptomics, metabolomics, proteomics, glycomics, bioinformatics, and computational microbiology. mSystems™ will provide streamlined decisions, while carrying on ASM''s tradition of rigorous peer review.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


