Unraveling the adsorption dynamics of asphaltene molecules on silica surfaces
IF 2.7
4区 生物学
Q2 BIOCHEMICAL RESEARCH METHODS
引用次数: 0
Abstract
Understanding the adsorption behavior of heavy oil components on reservoir solids is crucial for improving oil recovery, yet the molecular mechanism remains unclear. This study used molecular dynamics simulations to explore the adsorption kinetics and thermodynamics of asphaltene molecules on silica surfaces. The adsorption process was divided into three stages: free, adsorption, and equilibrium. In the adsorption stage, asphaltenes must pass through two dense hydration layers and adhere to the silica surface in a flat configuration. Carboxyl groups increase asphaltene hydrophilicity, raising interaction energy with water molecules and hindering adsorption. In addition, two distinct hydration layers of water molecules on the silica surface. The first hydration layer, with a peak density of 2000 kg m−3, was located around 0.6 nm from the surface, driven by hydrogen bonding between Si-OH groups and water molecules. The second layer, found at 1.44–1.80 nm, had a lower density of 1200 kg m−3, formed through hydrogen bonding between water molecules. This study aims to enhance the understanding of the physicochemical mechanisms governing oil droplet adsorption on silica surfaces, potentially informing the design of improved oil recovery strategies.
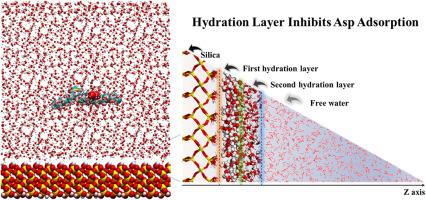
揭示沥青分子在二氧化硅表面的吸附动力学。
了解重油成分在储层固体上的吸附行为对提高石油采收率至关重要,但其分子机理仍不清楚。本研究利用分子动力学模拟来探索沥青质分子在二氧化硅表面的吸附动力学和热力学。吸附过程分为三个阶段:自由阶段、吸附阶段和平衡阶段。在吸附阶段,沥青质必须穿过两个致密的水合层,以平面构型附着在二氧化硅表面。羧基增加了沥青质的亲水性,提高了与水分子的相互作用能,阻碍了吸附。此外,二氧化硅表面有两个不同的水分子水合层。第一层水合层的峰值密度为 2000 kg m-3,位于距离表面 0.6 nm 左右的位置,由 Si-OH 基团和水分子之间的氢键驱动。第二层位于 1.44-1.80 纳米处,密度较低,为 1200 kg m-3,由水分子之间的氢键形成。这项研究旨在加深人们对硅表面吸附油滴的物理化学机制的了解,从而为改进采油战略的设计提供潜在的信息。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Journal of molecular graphics & modelling
生物-计算机:跨学科应用
CiteScore
5.50
自引率
6.90%
发文量
216
审稿时长
35 days
期刊介绍:
The Journal of Molecular Graphics and Modelling is devoted to the publication of papers on the uses of computers in theoretical investigations of molecular structure, function, interaction, and design. The scope of the journal includes all aspects of molecular modeling and computational chemistry, including, for instance, the study of molecular shape and properties, molecular simulations, protein and polymer engineering, drug design, materials design, structure-activity and structure-property relationships, database mining, and compound library design.
As a primary research journal, JMGM seeks to bring new knowledge to the attention of our readers. As such, submissions to the journal need to not only report results, but must draw conclusions and explore implications of the work presented. Authors are strongly encouraged to bear this in mind when preparing manuscripts. Routine applications of standard modelling approaches, providing only very limited new scientific insight, will not meet our criteria for publication. Reproducibility of reported calculations is an important issue. Wherever possible, we urge authors to enhance their papers with Supplementary Data, for example, in QSAR studies machine-readable versions of molecular datasets or in the development of new force-field parameters versions of the topology and force field parameter files. Routine applications of existing methods that do not lead to genuinely new insight will not be considered.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


