Drug–target prediction through self supervised learning with dual task ensemble approach
IF 2.6
4区 生物学
Q2 BIOLOGY
引用次数: 0
Abstract
Drug–Target interaction (DTI) prediction, a transformative approach in pharmaceutical research, seeks novel therapeutic applications for computational method based virtual screening, existing drugs to address untreated diseases and discovery of existing drugs side effects. The proposed model predict DTI through Heterogeneous biological network by combining drug, genes and disease related knowledge. For the purpose of embedding extraction Self-supervised learning (SSL) has been used which, trains models through pretext tasks, eliminating the need for manual annotations. The pretext tasks are related to either structural based information or similarity based information. To mitigate GNN vulnerability to non-robustness, ensemble learning can be incorporated into GNNs, harnessing multiple models to enhance robustness. This paper introduces a Graph neural network based architecture consisting of task based module and ensemble module for link prediction of DTI. The ensemble module of dual task combinations, both in cold start and warm start scenarios achieve very good performance as it provide 0.960 in cold start and 0.970 in warm start mean AUCROC score with less deviation.
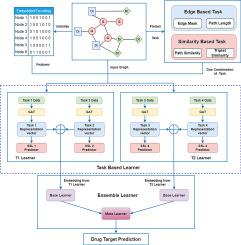
利用双任务集合方法,通过自我监督学习进行药物靶标预测。
药物-靶点相互作用(DTI)预测是制药研究中的一种变革性方法,它为基于计算方法的虚拟筛选、解决未治疗疾病的现有药物以及发现现有药物的副作用寻求新的治疗应用。所提出的模型结合了药物、基因和疾病相关知识,通过异构生物网络预测 DTI。为实现嵌入提取的目的,使用了自我监督学习(SSL),通过借口任务训练模型,从而消除了人工注释的需要。借口任务与基于结构的信息或基于相似性的信息有关。为了减轻图神经网络的不稳定性,可以将集合学习纳入图神经网络,利用多个模型来增强稳健性。本文介绍了一种基于图神经网络的架构,该架构由基于任务的模块和用于 DTI 链接预测的集合模块组成。双任务组合的集合模块在冷启动和暖启动情况下都取得了非常好的性能,冷启动平均 AUCROC 得分为 0.960,暖启动平均 AUCROC 得分为 0.970,且偏差较小。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational Biology and Chemistry
生物-计算机:跨学科应用
CiteScore
6.10
自引率
3.20%
发文量
142
审稿时长
24 days
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


