Cycle-ESM: Generation-assisted classification of antifungal peptides using ESM protein language model
IF 2.6
4区 生物学
Q2 BIOLOGY
引用次数: 0
Abstract
The rising prevalence of invasive fungal infections and the emergence of antifungal resistance highlight the urgent need for new antifungal medications. Antifungal peptides have emerged as promising alternatives to traditional antimicrobial agents. The identification of natural or synthetic antifungal peptides is crucial for advancing antifungal drug development. Typically, the availability of antifungal samples is limited, and significant sequence diversity exists among antifungal peptides, posing challenges for high-throughput screening. To address the identification challenge of antifungal peptides with limited sample availability, this study introduces the Cycle ESM method. Initially, the method utilises the ESM protein language model to generate additional data on antifungal peptides, serving as a data augmentation technique to enhance model training effectiveness. Subsequently, the ESM is employed in conjunction with a textCNN model to construct a classifier for peptide prediction, with a comprehensive exploration of peptide characteristics to improve prediction accuracy. Experimental results demonstrate that the performance of the Cycle ESM method surpasses that of existing methods across three distinct antifungal peptide datasets. This study presents a novel approach to antifungal peptide prediction and offers innovative insights for addressing classification problems with limited sample availability.
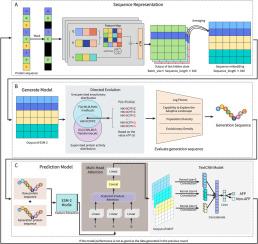
Cycle-ESM:使用 ESM 蛋白语言模型对抗真菌肽进行世代辅助分类。
随着侵袭性真菌感染发病率的上升和抗真菌耐药性的出现,迫切需要新的抗真菌药物。抗真菌肽作为传统抗菌剂的替代品已经崭露头角。天然或合成抗真菌肽的鉴定对于推动抗真菌药物的开发至关重要。通常情况下,抗真菌样本的可用性有限,而且抗真菌肽之间存在显著的序列多样性,这给高通量筛选带来了挑战。为了解决在样品有限的情况下鉴定抗真菌肽的难题,本研究引入了循环 ESM 方法。首先,该方法利用 ESM 蛋白语言模型生成额外的抗真菌肽数据,作为一种数据增强技术来提高模型训练的有效性。随后,ESM 与 textCNN 模型结合使用,构建肽预测分类器,全面探索肽的特征,提高预测准确性。实验结果表明,在三个不同的抗真菌肽数据集上,周期ESM方法的性能超过了现有方法。这项研究提出了一种新的抗真菌肽预测方法,为解决样本有限的情况下的分类问题提供了创新见解。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Computational Biology and Chemistry
生物-计算机:跨学科应用
CiteScore
6.10
自引率
3.20%
发文量
142
审稿时长
24 days
期刊介绍:
Computational Biology and Chemistry publishes original research papers and review articles in all areas of computational life sciences. High quality research contributions with a major computational component in the areas of nucleic acid and protein sequence research, molecular evolution, molecular genetics (functional genomics and proteomics), theory and practice of either biology-specific or chemical-biology-specific modeling, and structural biology of nucleic acids and proteins are particularly welcome. Exceptionally high quality research work in bioinformatics, systems biology, ecology, computational pharmacology, metabolism, biomedical engineering, epidemiology, and statistical genetics will also be considered.
Given their inherent uncertainty, protein modeling and molecular docking studies should be thoroughly validated. In the absence of experimental results for validation, the use of molecular dynamics simulations along with detailed free energy calculations, for example, should be used as complementary techniques to support the major conclusions. Submissions of premature modeling exercises without additional biological insights will not be considered.
Review articles will generally be commissioned by the editors and should not be submitted to the journal without explicit invitation. However prospective authors are welcome to send a brief (one to three pages) synopsis, which will be evaluated by the editors.
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


