CHK1 attenuates cardiac dysfunction via suppressing SIRT1-ubiquitination
IF 10.8
1区 医学
Q1 ENDOCRINOLOGY & METABOLISM
引用次数: 0
Abstract
Background
Mitochondrial dysfunction is linked to myocardial ischemia-reperfusion (I/R) injury. Checkpoint kinase 1 (CHK1) could facilitate cardiomyocyte proliferation, however, its role on mitochondrial function in I/R injury remains unknown.
Methods
To investigate the role of CHK1 on mitochondrial function following I/R injury, cardiomyocyte-specific knockout/overexpression mouse models were generated. Adult mouse cardiomyocytes (AMCMs) were isolated for in vitro study. Mass spectrometry-proteomics analysis and protein co-immunoprecipitation assays were conducted to dissect the molecular mechanism.
Results
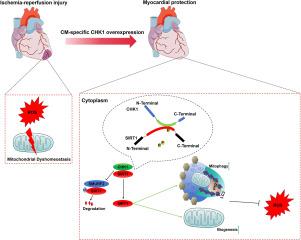
CHK1 was downregulated in myocardium post I/R and AMCMs post oxygen-glucose deprivation/re‑oxygenation (OGD/R). In vivo, CHK1 overexpression protected against I/R induced cardiac dysfunction, while heterogenous CHK1 knockout exacerbated cardiomyopathy. In vitro, CHK1 inhibited OGD/R-induced cardiomyocyte apoptosis and bolstered cardiomyocyte survival. Mechanistically, CHK1 attenuated oxidative stress and preserved mitochondrial metabolism in cardiomyocytes under I/R. Moreover, disrupted mitochondrial homeostasis in I/R myocardium was restored by CHK1 through the promotion of mitochondrial biogenesis and mitophagy. Through mass spectrometry analysis following co-immunoprecipitation, SIRT1 was identified as a direct target of CHK1. The 266–390 domain of CHK1 interacted with the 160–583 domain of SIRT1. Importantly, CHK1 phosphorylated SIRT1 at Thr530 residue, thereby inhibiting SMURF2-mediated degradation of SIRT1. The role of CHK1 in maintaining mitochondrial dynamics control and myocardial protection is abolished by SIRT1 inhibition, while inactivated mutation of SIRT1 Thr530 fails to reverse the impaired mitochondrial dynamics following CHK1 knockdown. CHK1 Δ390 amino acids (aa) mutant functioned similarly to full-length CHK1 in scavenging ROS and maintaining mitochondrial dynamics. Consistently, cardiac-specific SIRT1 knockdown attenuated the protective role of CHK1 in I/R injury.
Conclusions
Our findings revealed that CHK1 mitigates I/R injury and restores mitochondrial dynamics in cardiomyocytes through a SIRT1-dependent mechanism.
CHK1 通过抑制 SIRT1 泛素化减轻心脏功能障碍
背景:线粒体功能障碍与心肌缺血再灌注(I/R)损伤有关。检查点激酶 1(CHK1)可促进心肌细胞增殖,但它在 I/R 损伤中对线粒体功能的作用仍不清楚:方法:为了研究 I/R 损伤后 CHK1 对线粒体功能的作用,我们建立了心肌细胞特异性基因敲除/表达小鼠模型。分离成年小鼠心肌细胞(AMCMs)进行体外研究。通过质谱-蛋白质组学分析和蛋白质共沉淀实验来揭示其分子机制:结果:CHK1在I/R后的心肌和氧-葡萄糖剥夺/再氧合(OGD/R)后的AMCMs中下调。在体内,CHK1 的过表达可防止 I/R 引起的心脏功能障碍,而异源 CHK1 敲除会加重心肌病。在体外,CHK1 可抑制 OGD/R 诱导的心肌细胞凋亡,提高心肌细胞存活率。从机理上讲,CHK1 可减轻氧化应激,保护 I/R 条件下心肌细胞的线粒体代谢。此外,CHK1 还通过促进线粒体生物生成和有丝分裂来恢复 I/R 心肌中被破坏的线粒体平衡。通过共免疫沉淀后的质谱分析,SIRT1 被确定为 CHK1 的直接靶标。CHK1 的 266-390 结构域与 SIRT1 的 160-583 结构域相互作用。重要的是,CHK1 在 Thr530 残基上磷酸化了 SIRT1,从而抑制了 SMURF2 介导的 SIRT1 降解。抑制 SIRT1 会取消 CHK1 在维持线粒体动力学控制和心肌保护方面的作用,而 SIRT1 Thr530 的失活突变无法逆转 CHK1 敲除后线粒体动力学受损的情况。CHK1 Δ390氨基酸(aa)突变体在清除ROS和维持线粒体动力学方面的功能与全长CHK1相似。同样,心脏特异性 SIRT1 基因敲除削弱了 CHK1 在 I/R 损伤中的保护作用:我们的研究结果表明,CHK1 可通过 SIRT1 依赖性机制减轻 I/R 损伤并恢复心肌细胞线粒体的活力。
本文章由计算机程序翻译,如有差异,请以英文原文为准。
求助全文
约1分钟内获得全文
求助全文
来源期刊

Metabolism: clinical and experimental
医学-内分泌学与代谢
CiteScore
18.90
自引率
3.10%
发文量
310
审稿时长
16 days
期刊介绍:
Metabolism upholds research excellence by disseminating high-quality original research, reviews, editorials, and commentaries covering all facets of human metabolism.
Consideration for publication in Metabolism extends to studies in humans, animal, and cellular models, with a particular emphasis on work demonstrating strong translational potential.
The journal addresses a range of topics, including:
- Energy Expenditure and Obesity
- Metabolic Syndrome, Prediabetes, and Diabetes
- Nutrition, Exercise, and the Environment
- Genetics and Genomics, Proteomics, and Metabolomics
- Carbohydrate, Lipid, and Protein Metabolism
- Endocrinology and Hypertension
- Mineral and Bone Metabolism
- Cardiovascular Diseases and Malignancies
- Inflammation in metabolism and immunometabolism
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


