Prashant Sharma, Jason R McFadden, F Graeme Frost, Thomas C Markello, Dorothy K Grange, Wendy J Introne, William A Gahl, May Christine V Malicdan
{"title":"Biallelic germline DDX41 variants in a patient with bone dysplasia, ichthyosis, and dysmorphic features.","authors":"Prashant Sharma, Jason R McFadden, F Graeme Frost, Thomas C Markello, Dorothy K Grange, Wendy J Introne, William A Gahl, May Christine V Malicdan","doi":"10.1007/s00439-024-02708-8","DOIUrl":null,"url":null,"abstract":"<p><p>DDX41 (DEAD‑box helicase 41) is a member of the largest family of RNA helicases. The DEAD-box RNA helicases share a highly conserved core structure and regulate all aspects of RNA metabolism. The functional role of DDX41 in innate immunity is also highly conserved. DDX41 acts as a sensor of viral DNA and activates the STING-TBK1-IRF3-type I IFN signaling pathway. Germline heterozygous variants in DDX41 have been reported in familial myelodysplasia syndrome (MDS)/acute myeloid leukemia (AML) patients; most patients also acquired a somatic variant in the second DDX41 allele. Here, we report a patient who inherited compound heterozygous DDX41 variants and presented with bone dysplasia, ichthyosis, and dysmorphic features. Functional analyses of the patient-derived dermal fibroblasts revealed a reduced abundance of DDX41 and abrogated activation of the IFN genes through the STING-type I interferon pathway. Genome-wide transcriptome analyses in the patient's fibroblasts revealed significant gene dysregulation and changes in the RNA splicing events. The patient's fibroblasts also displayed upregulation of periostin mRNA expression. Using an RNA binding protein assay, we identified DDX41 as a novel regulator of periostin expression. Our results suggest that functional impairment of DDX41, along with dysregulated periostin expression, likely contributes to this patient's multisystem disorder.</p>","PeriodicalId":13175,"journal":{"name":"Human Genetics","volume":" ","pages":"1445-1457"},"PeriodicalIF":3.6000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11576897/pdf/","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Human Genetics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1007/s00439-024-02708-8","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/25 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
Abstract
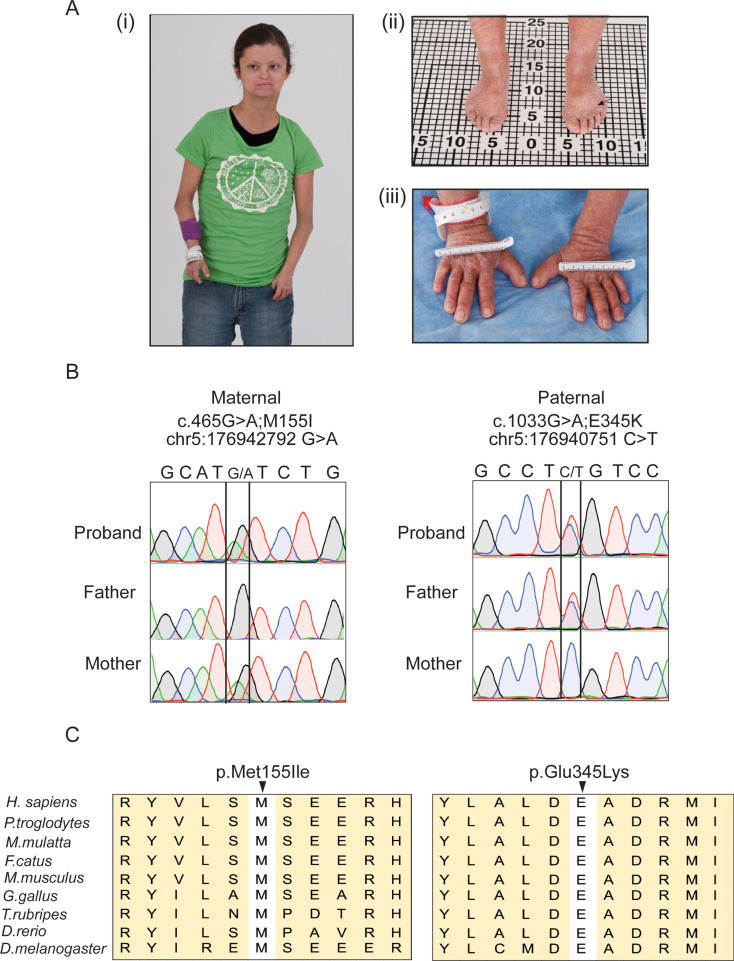
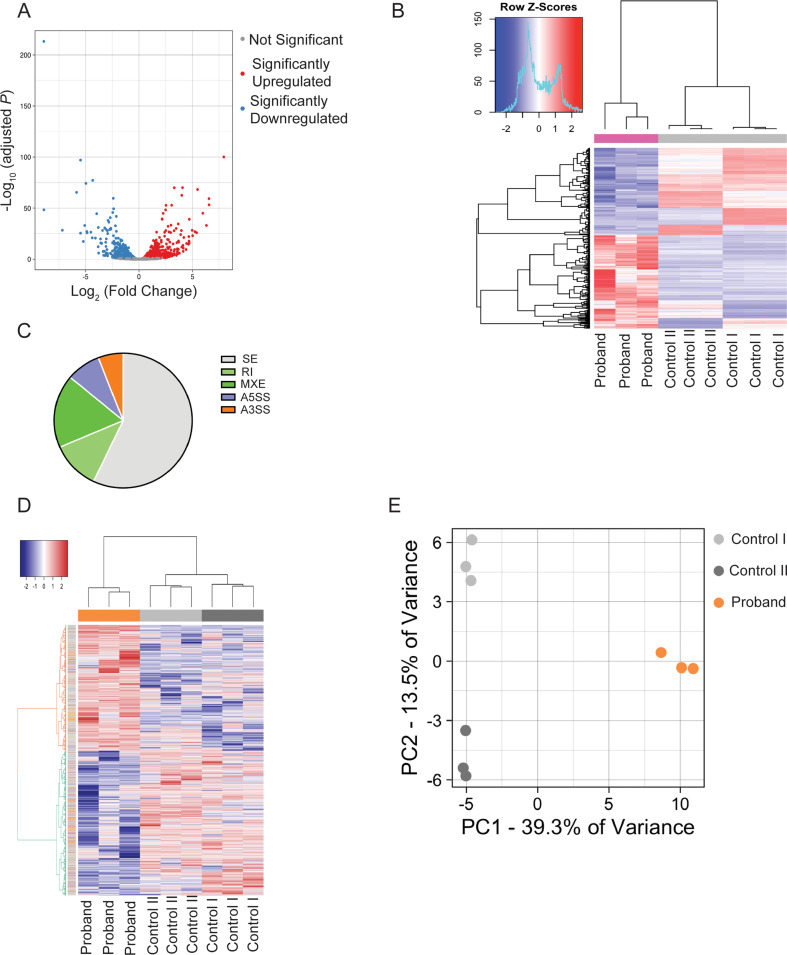
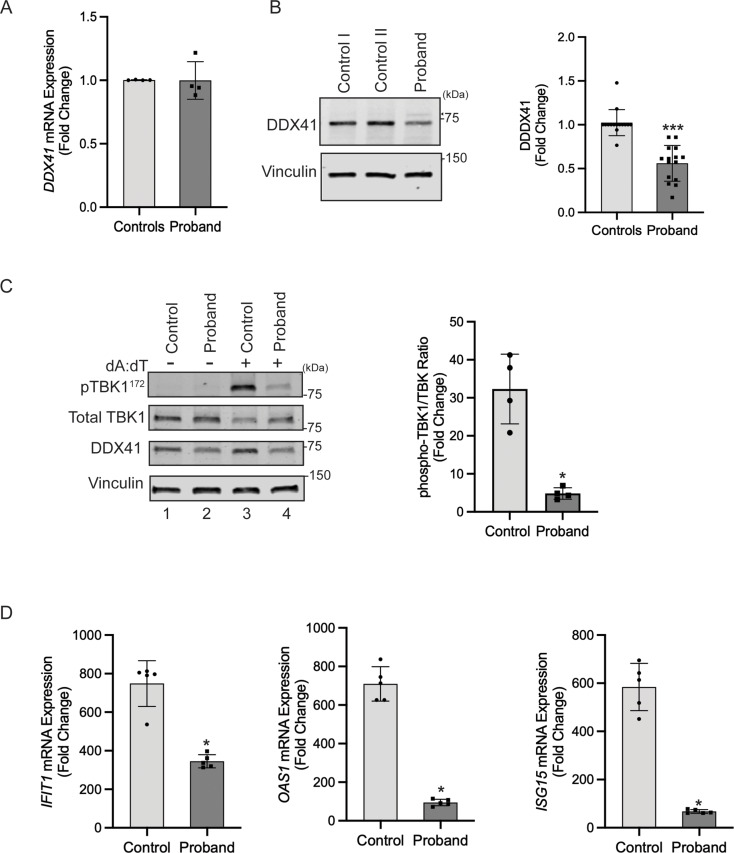
DDX41 (DEAD‑box helicase 41) is a member of the largest family of RNA helicases. The DEAD-box RNA helicases share a highly conserved core structure and regulate all aspects of RNA metabolism. The functional role of DDX41 in innate immunity is also highly conserved. DDX41 acts as a sensor of viral DNA and activates the STING-TBK1-IRF3-type I IFN signaling pathway. Germline heterozygous variants in DDX41 have been reported in familial myelodysplasia syndrome (MDS)/acute myeloid leukemia (AML) patients; most patients also acquired a somatic variant in the second DDX41 allele. Here, we report a patient who inherited compound heterozygous DDX41 variants and presented with bone dysplasia, ichthyosis, and dysmorphic features. Functional analyses of the patient-derived dermal fibroblasts revealed a reduced abundance of DDX41 and abrogated activation of the IFN genes through the STING-type I interferon pathway. Genome-wide transcriptome analyses in the patient's fibroblasts revealed significant gene dysregulation and changes in the RNA splicing events. The patient's fibroblasts also displayed upregulation of periostin mRNA expression. Using an RNA binding protein assay, we identified DDX41 as a novel regulator of periostin expression. Our results suggest that functional impairment of DDX41, along with dysregulated periostin expression, likely contributes to this patient's multisystem disorder.
期刊介绍:
Human Genetics is a monthly journal publishing original and timely articles on all aspects of human genetics. The Journal particularly welcomes articles in the areas of Behavioral genetics, Bioinformatics, Cancer genetics and genomics, Cytogenetics, Developmental genetics, Disease association studies, Dysmorphology, ELSI (ethical, legal and social issues), Evolutionary genetics, Gene expression, Gene structure and organization, Genetics of complex diseases and epistatic interactions, Genetic epidemiology, Genome biology, Genome structure and organization, Genotype-phenotype relationships, Human Genomics, Immunogenetics and genomics, Linkage analysis and genetic mapping, Methods in Statistical Genetics, Molecular diagnostics, Mutation detection and analysis, Neurogenetics, Physical mapping and Population Genetics. Articles reporting animal models relevant to human biology or disease are also welcome. Preference will be given to those articles which address clinically relevant questions or which provide new insights into human biology.
Unless reporting entirely novel and unusual aspects of a topic, clinical case reports, cytogenetic case reports, papers on descriptive population genetics, articles dealing with the frequency of polymorphisms or additional mutations within genes in which numerous lesions have already been described, and papers that report meta-analyses of previously published datasets will normally not be accepted.
The Journal typically will not consider for publication manuscripts that report merely the isolation, map position, structure, and tissue expression profile of a gene of unknown function unless the gene is of particular interest or is a candidate gene involved in a human trait or disorder.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


