{"title":"Theoretical exploration of energetic molecular design strategy: functionalization of C or N and structural selection of imidazole or pyrazole","authors":"Qianxiong Chen, Jin Zhu, Suming Jing, Jiahao Deng, Yuanyuan Wang, Keyao Li, Zhineng Wang, Jia Liu, Shuai Bian","doi":"10.1007/s00894-024-06183-w","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>In researching energetic materials with high energy density, it is an effective method to introduce explosophoric groups. In this study, four series of energetic compounds were designed by functionalizing with C- or N-, introducing energetic groups -CH(NO<sub>2</sub>)<sub>2</sub>, -CF(NO<sub>2</sub>)<sub>2</sub>, -C(NO<sub>2</sub>)<sub>2</sub>(NF<sub>2</sub>), -C(NO<sub>2</sub>)<sub>3</sub>, and-CH(NF<sub>2</sub>)<sub>2</sub> into imidazole and pyrazole structures. Density functional theory was employed to optimize the structure of the target compound and subsequently to predict and evaluate its performance based on this. Meanwhile, the sensitivity of the compounds was predicted based on their electrostatic potential analysis. Following analysis of the geometric structure, detonation performance, and sensitivity of the compounds, three factors were discussed: energetic groups, functionalization methods, and skeleton structure differences. The results indicate that C-functionalization has advantages only in density, but N-functionalization is better in thermal stability, heat of formation, and sensitivity. Meanwhile, the data shows that imidazole-based compounds exhibited greater density and detonation performance in the target compounds designed within this study, while pyrazoles have a higher heat of formation and chemical stability. By analyzing the design strategy of C- or N-functionalization of novel high-energy groups on energetic imidazole or pyrazole rings and selecting a more suitable molecular construction strategy, this study provides a theoretical approach for the development of new energetic materials with excellent performance.</p><h3>Method</h3><p>Gaussian 09 and Multiwfn 3.8 packages are the software used for calculation, and the electrostatic potentials were depicted using the VMD program. In this study, the imidazole and pyrazole derivatives were optimized at the B3PW91/6-311G (d, p) level to acquire the relevant data for the compounds.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":null,"pages":null},"PeriodicalIF":2.1000,"publicationDate":"2024-10-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06183-w","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
Abstract
Context
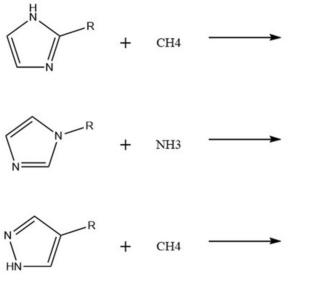
In researching energetic materials with high energy density, it is an effective method to introduce explosophoric groups. In this study, four series of energetic compounds were designed by functionalizing with C- or N-, introducing energetic groups -CH(NO2)2, -CF(NO2)2, -C(NO2)2(NF2), -C(NO2)3, and-CH(NF2)2 into imidazole and pyrazole structures. Density functional theory was employed to optimize the structure of the target compound and subsequently to predict and evaluate its performance based on this. Meanwhile, the sensitivity of the compounds was predicted based on their electrostatic potential analysis. Following analysis of the geometric structure, detonation performance, and sensitivity of the compounds, three factors were discussed: energetic groups, functionalization methods, and skeleton structure differences. The results indicate that C-functionalization has advantages only in density, but N-functionalization is better in thermal stability, heat of formation, and sensitivity. Meanwhile, the data shows that imidazole-based compounds exhibited greater density and detonation performance in the target compounds designed within this study, while pyrazoles have a higher heat of formation and chemical stability. By analyzing the design strategy of C- or N-functionalization of novel high-energy groups on energetic imidazole or pyrazole rings and selecting a more suitable molecular construction strategy, this study provides a theoretical approach for the development of new energetic materials with excellent performance.
Method
Gaussian 09 and Multiwfn 3.8 packages are the software used for calculation, and the electrostatic potentials were depicted using the VMD program. In this study, the imidazole and pyrazole derivatives were optimized at the B3PW91/6-311G (d, p) level to acquire the relevant data for the compounds.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式:


